键合、结构和共振
杂化轨道和杂化
最后更新:2022年12月13日|
今天的帖子是关于杂化轨道.以下是一个简短的总结:
[注:本文与Matthew Pierce共同撰写有机化学解决方案.问问马特安排在线辅导课程的事在这里.]
目录
- 甲烷碳周围碳氢键的四面体排列中涉及哪些轨道(CH4)?
- 杂化轨道:碳原子成键的解释
- 流行的比喻:混合汽水
- sp3.杂化准确地描述了与四个原子键合的(第一排)元素中的原子排列…
- ……以及[原子+孤对]数量等于4的情况
- sp2杂交
- sp杂化
- 总结-杂化轨道
- 笔记
1.甲烷的四面体排列中涉及哪些轨道(CH4)?
在上一篇关于甲烷结构的文章中,我们问我们是如何知道甲烷是四面体的(见文章:我们怎么知道甲烷是四面体).
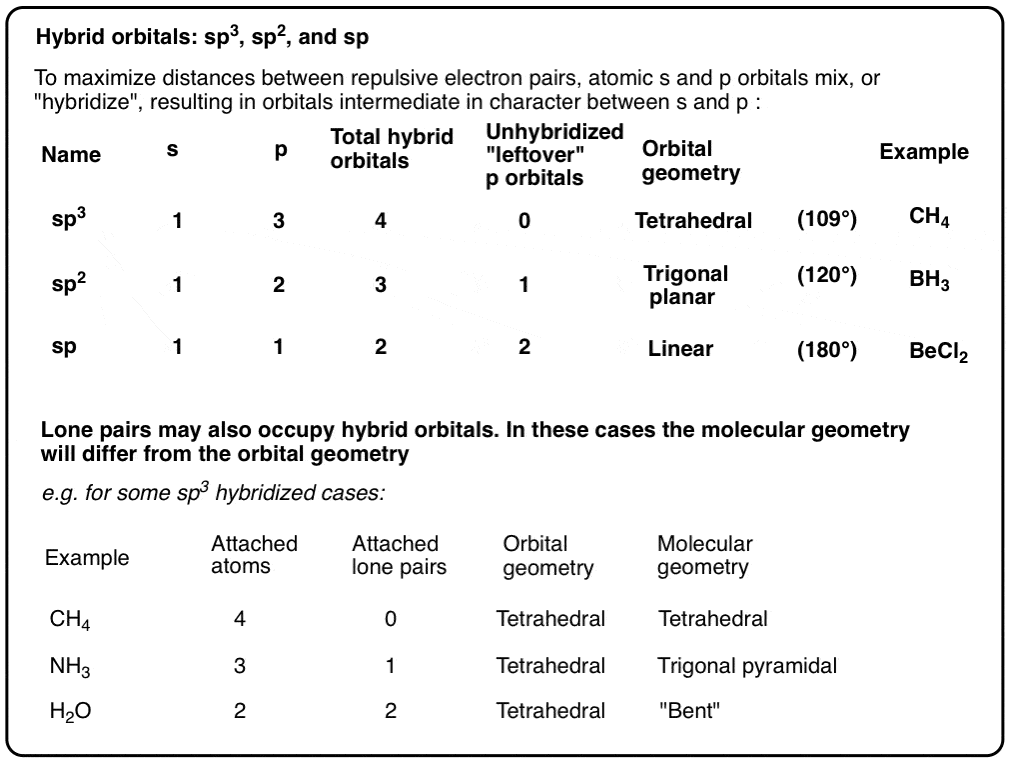
基于碳的轨道(2s和2p轨道),我们可能天真地期望其中三个碳氢键分别沿x、y和z轴排列,而另一个键在某个任意角度(135°左右)
但是,就像在科学中经常发生的那样,我们美丽的直觉假设被一些恼人的实验事实摧毁了:
- 甲烷没有可测量的偶极矩(我们的“合理”结构,左下方,将预期具有可测量的偶极矩)
- 金刚石的晶体结构是四面体,碳原子之间的键长和角相同,为109.5度。这不是我们所期望的,如果我们处理的是“纯”2s和2p轨道之间的键!
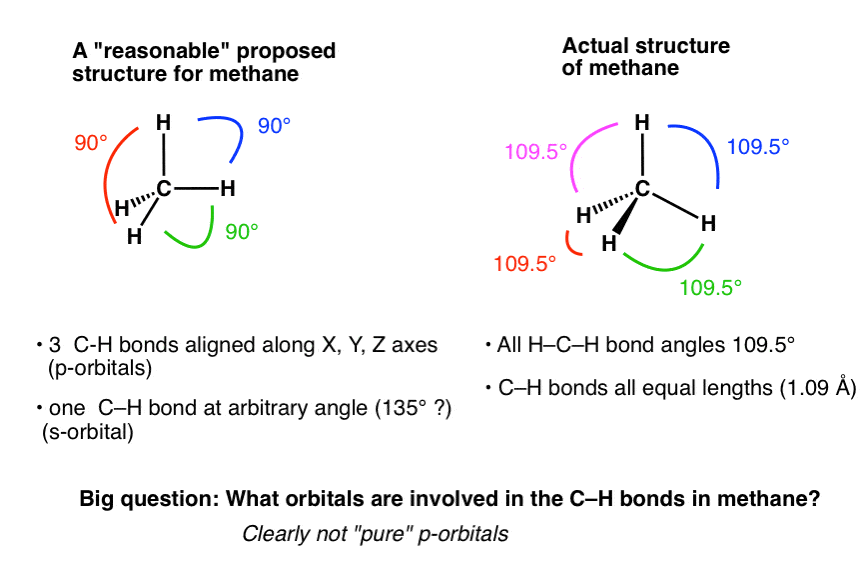
回想起来,几何图形是有意义的。恰好109.5度是使四个键对之间的距离最大化的方向,从而使它们的排斥性相互作用最小化。换句话说,几何是直接的结果的“异性电荷相吸,同性电荷相斥”。
但是我们如何描述轨道是用来给出键角的吗?
这真是件令人头痛的事。它们不可能是纯2s轨道(反正只有一个2s轨道)它们不可能是纯p轨道,因为p轨道是90°的。
这是什么样的轨道是他们吗?
2.杂化轨道:碳原子成键的解释
莱纳斯·鲍林在他的经典著作《化学键的性质》(1931)中提出了同样的问题[pdf这在很大程度上为他赢得了1954年诺贝尔化学奖.
鲍林对这一困境的解决方案,我们今天仍然适用,是这样的:
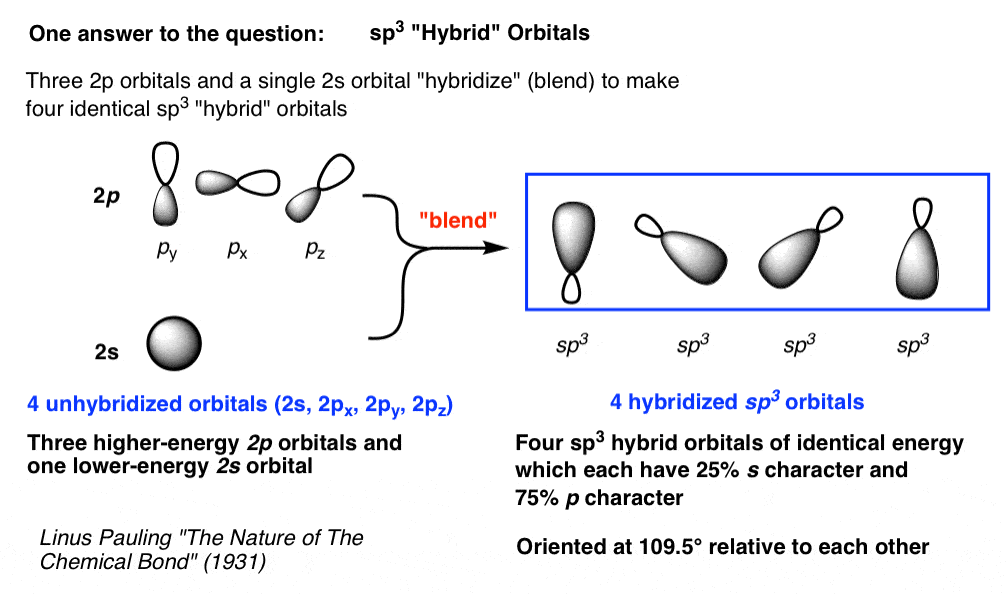
- 甲烷中的成键轨道没有一个是100%的s轨道或100%的p轨道,相反,它们是杂化轨道每个都有部分年代特征和部分p的性格。
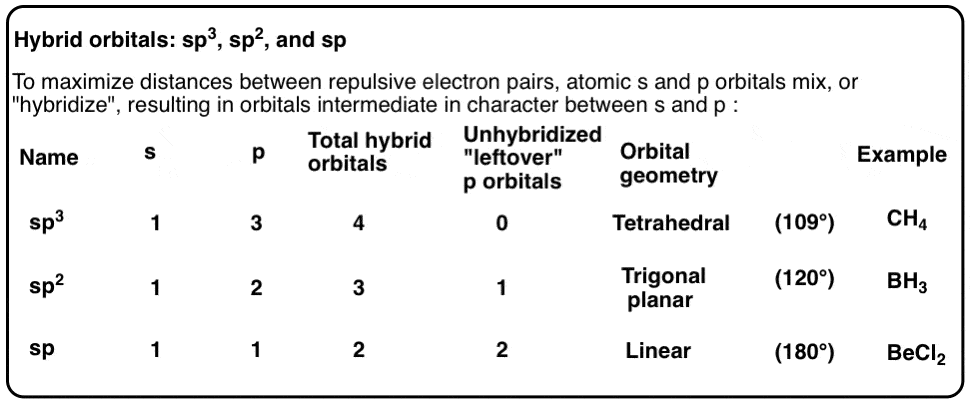
- 三个2p轨道和一个2s轨道杂交(即混合)以创建四个混合sp3.轨道,围绕中心碳原子四面体排列。
- 四个杂化轨道都有25%的s轨道和75%的p轨道。
- 以甲烷为例,每一个sp3.杂化轨道与氢的1s轨道重叠形成碳氢键
我在这里实话实说。许多学生不喜欢这种解释。
当然,一开始会让人困惑。为什么?
我想当我们最初学习轨道的时候,我们本能地认为它们是容器有点像原子特百惠,用来装电子,而电子恰好有各种可爱的形状。

因此,可以想象电子的行为就像在这些容器中叮当作响的“水果”(严格的限制:每个集装箱2个!).当我们知道它们可以在一个原子中从一个容器移动到另一个容器,甚至完全离开原子时,我们的直觉并不奇怪。
什么是令人惊讶的是,当我们打开冰箱,发现我们可爱的球形和哑铃状的特百惠容器改变他们的形状把他们的财产合并了!这违背了我们对容器行为的直觉!
{kind=link}

伙计们,这是量子世界!如果这不让你迷惑…该死的应该!
今后,我不要求你们理解,甚至不要求你们相信杂交在深刻的数学或理论层面上。这对于我们的目的来说是不必要的[鲍林是一位伟大的老师。你可以阅读鲍林的原始论文,在这里,如果你愿意]
我只是建议你试一试放下你的怀疑,因为用这个杂交模型将帮助我们合理化很多分子的结构,几何和行为。
3.流行的比喻:混合汽水
在我们进一步深入之前,这里有一个我认为有用的小类比(谢谢,史蒂文),这可能有助于让人明白这一点。
- 假设你有四瓶汽水:一瓶雪碧(Sprite)和三瓶百事可乐(Pepsi)。
- 现在想象一下,把它们倒出来,混合在一起,然后把混合物重新装进每个瓶子。
- 波普守恒定律说你还有四瓶液体。但现在的流行既不是纯雪碧,也不是纯百事;它是两者的混合体。
- 具体来说,每个瓶子现在有25%的雪碧角色和75%的百事可乐角色。
- 我们可以叫它"混合"流行音乐,如果你喜欢,也可以叫sp3.
![]()
这有点像2s和2p轨道的情况。通过混合2s轨道和3个2p轨道,我们现在有4个轨道有25%的s轨道和75%的p轨道。(尽管重要的是,瓶子的形状在变化,而不仅仅是内容)。
最后一步是在四面体的四角上排列这四个轨道,这允许四个电子对之间的最大距离(同性电荷相斥!)
这种合理化对我们有什么帮助呢?
- 它解释了甲烷的四面体分子几何结构,具有4个相同的H-C-H键角(109.5°)
- 它解释了甲烷中4个相同的碳氢键长度(和键强度)
- 它解释了甲烷中偶极矩的缺乏,因为电子对的四面体排列允许所有部分电荷抵消(即向量和为零)
- 这个模型甚至有助于解释某些反应是如何发生的,我们现在不会深入讨论[例如,在SN2个反应发生在空的“反键”轨道上,与成键的C-H轨道180°,如果你之前读过的话]
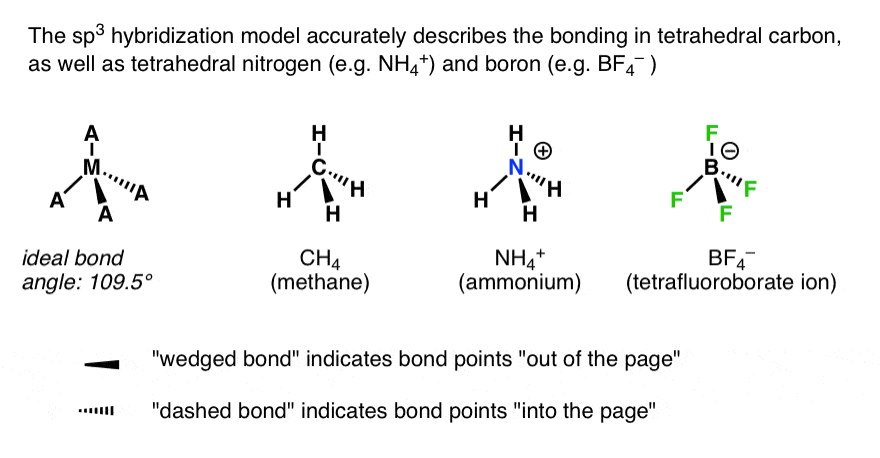
4.sp3.杂化准确地描述了与四个原子键合的(第一排)元素中的原子排列…
这不仅适用于甲烷。它适用于任何一个(第一行)元素与四个原子成键的情况。显然,四面体碳是我们将要探讨的最突出的例子,但它也适用于四面体氮[例如NH]4(+)]和甚至四面体硼[例如BF4(-)]。
5……以及[原子+孤对]数等于4的情况
轨道的四面体排列是成立的即使有一对或多对电子是未成键的孤对(如果你遇到了VSEPR理论大多数人在学习有机化学的时候都有,这并不令人惊讶.)
例如,氨(NH3.),即“甲基阴离子”(CH3.-)及水合氢离子(H3.O+)都有一个中心原子,有4对电子:3对成键电子和一对非成键电子。正如我们所看到的,排列四对电子的理想几何结构是四面体,这使得杂交中心原子sp3..这样分子就形成了围绕中心原子的“钢琴凳”排列,我们称之为“三角金字塔”几何结构。
另一种说法是,中心原子是四面体轨道几何(sp3.)和三角平面分子几何。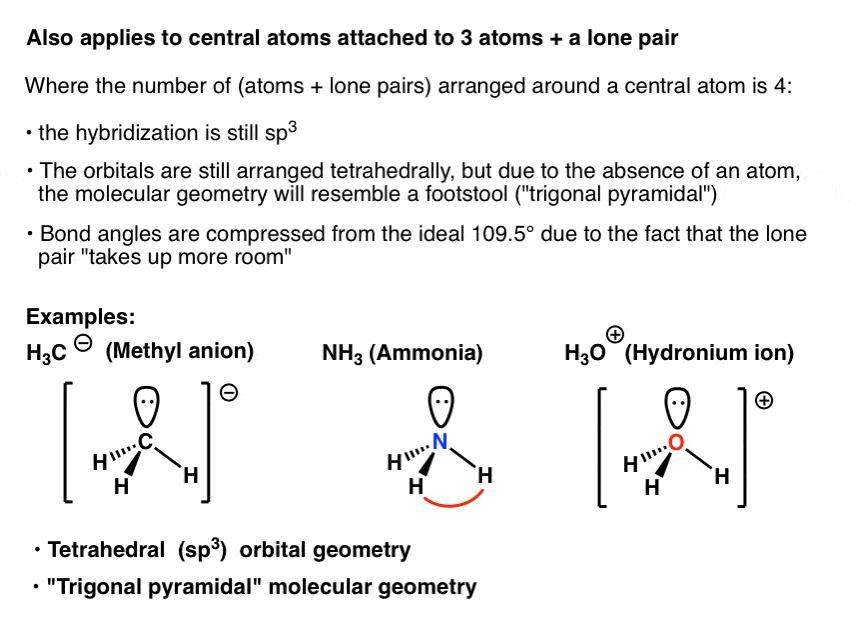
一个有趣的事实是,其中的键角是从109.5°的理想角度压缩的。例如,氨中的H-N-H键角是107度。我们认为这是由于非成键的孤对比“正常”成键对具有更强的排斥性——可能是因为它更接近原子并施加更强的影响。
这种偏离“理想”键角在H中甚至更大2O(水)有两个孤对。的杂交仍然是sp3.,轨道几何仍然是四面体,但所得到的分子的形状(“分子几何”)是“弯曲的”。

的确,这是鲍林的显著成就之一杂交模型是,它正确地解释了水的偶极矩.如果水是完美的“线性”,就像我们许多人在学习化学之前天真地认为的那样,偶极子会相互抵消。
另一个“弯曲”几何的例子是酰胺阴离子NH2- - - - - -氮上有两个孤对。
一个快速的表格可能有助于总结我们已经建立的关于sp的一切3.杂交:

6.sp2杂交
让我们回到汽水瓶的类比。假设我们只把雪碧和两瓶百事可乐混合,而不是三瓶。
这给我们留下了什么?
![]()
这给了我们三个混合瓶装汽水,还有一个剩下的unhybridized瓶子。
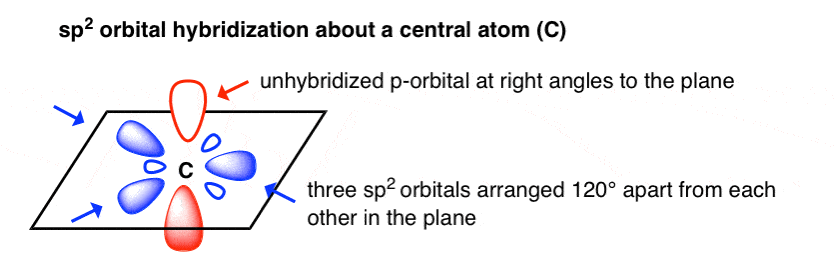
通过类比,如果我们把2s轨道和两个2p轨道混合,我们得到三个sp2杂化轨道,和一个剩下的,未杂化的p轨道。
当这三个2轨道充满电子对,使它们之间距离最大的键角是120°。
这给了我们一个sp的“三角平面”排列2轨道,未杂化的p轨道与平面成直角。有点像奔驰的标志。
三角形平面几何的一个经典例子是硼烷,BH3.它有三对成键电子,彼此呈120°排列。三角平面几何也发现在碳正离子,如甲基阳离子,CH3.+.

你可能会想,未杂化的p轨道在哪?
与平面成直角。回想一下,三个p轨道都是直角的。所以无论哪两个p轨道杂化,第三个(剩下的)p轨道将与它们形成的平面成直角(就像z轴垂直于xy平面一样)
以BH为例3.和碳正离子,未杂化的p轨道是空的。
还有一种很常见的情况,sp2然而,几何学是可以观察到的。如果相邻原子在未杂化的p轨道上有单电子,如果这些p轨道可以重叠,就可以形成键。这种现象被称为"pi-bonding”。(关于这一点,我们稍后会有更多的讨论。)
π键通常被称为“双键”,它需要一个未杂化的p轨道才能形成。
下面例子中的碳,氧和氮原子,它们都有π键(双键)是sp2杂化。轨道以大约120°的角度分开。
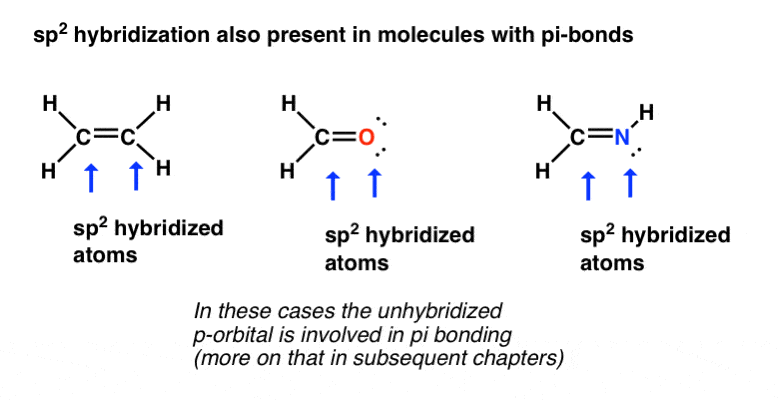
注意孤对可以在sp中2-杂化轨道,就像我们在NH中看到的3.和H2O在sp的情况下3.杂交.还要注意,在中间分子(甲醛)中,氧有两个孤对(每个以sp2杂化轨道)在右上方的分子中氮分子中有一个孤对2杂化轨道。
当孤对存在时,键角将略小于120°,因为孤对可以被认为占据了更多的“空间”。
7.sp杂化
让我们看看最后一种可能的情况。如果只有一个p轨道和s轨道杂化呢?
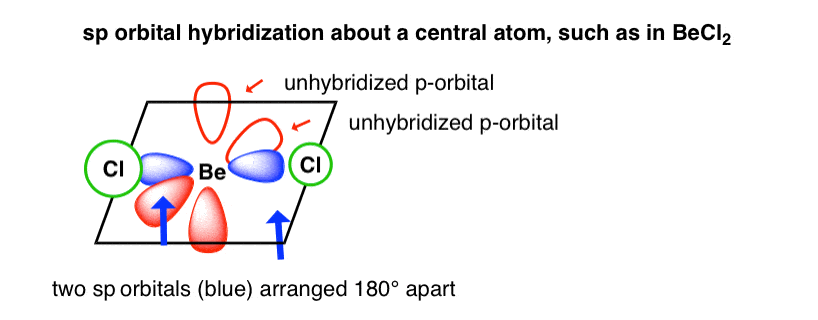
这就得到了两个杂化的“sp”轨道,它们之间的最大夹角是180度。我们称这种排列为“线性”。每个杂化sp轨道都有50%的s和50%的p。
![]()
两个未杂化的p轨道都与sp杂化轨道成直角。
例如,这是氯化铍(BeCl)的轨道2),其中Cl-Be-Cl键角为180°。如果我们认为Cl-Be-Cl键沿x轴,那么两个(未杂化的)p轨道将分别沿y轴和z轴。
sp -杂交比较常见的情况是两个成键在单个原子上.最突出的例子是“三键”,如图所示炔烃,腈,以及一氧化碳。在这些情况下,不仅碳原子是sp杂化的,而且氮原子也是sp杂化的腈)和氧原子(在一氧化碳中)。
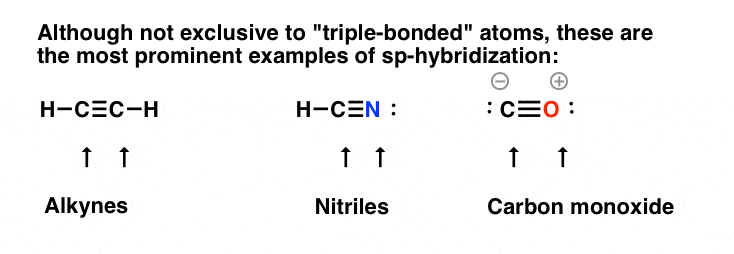
还要注意孤对可以在sp杂化轨道中,如图所示腈还有一氧化碳。
sp -杂交并非只有三键原子才有,例如,烯和烯酮的中心碳参与两个PI键,因此是sp杂化的,但三键是最容易识别的例子。
8.总结-杂化轨道
好的。以下是这篇无意中写得很长的文章的要点:
- 电子对相互排斥。在没有轨道时杂交,围绕CH的键角4会被限制在p轨道的几何形状(90°)它在能量上对s和p轨道有利杂交形成sp3.导致电子对更大的分离,键角为109°(即在四面体的顶点)。这也适用于具有非成键电子对的中心原子,如NH3.和H2O。
- 当只有两个p轨道参与时杂交,三个sp2杂化轨道采用三角平面轨道几何。剩下的(未杂化的)p轨道,与三角平面成直角,可以是空的(如在BH3.)或单占据(如在含有PI键的分子中所见)。
- 只有一个p轨道参与杂交,结果是两个sp杂化轨道和一个线性轨道几何。剩下的两个p轨道可用于成pi键(如三键有机化合物,如炔烃而且腈)或可以为空(如在BeCl中2).
- 注意围绕中心原子的轨道数是总是4.轨道既不会被创造也不会被破坏杂交;它们只是被转化了。
在下一篇文章中,我们将提供一个超级简单的技巧来快速确定杂交中心原子的。
再次感谢Matt的合作。问问马特安排在线辅导课程的事在这里.
感谢James(还有Matt)的另一篇非常有说服力的文章。
另一件一直困扰我的事情,也是与这个话题有关的,是氧的杂化(尤其是解释水的键角为104.5°)。
我和很多人和教授谈过,似乎在这个问题上的意见很分歧。
一方面,有人说氧的杂化就像碳的杂化一样,两个孤对将角从109.5°推到104.5°。
另一方面,人们声称氧的s轨道和p轨道在能量上距离太远,无法杂化,这个模型只是一个简单但不准确的解释。
根据这一点,两个氢以90°的角度结合到两个p轨道上,但由于斥力,它们并不完全位于p轴上,而是略微偏离(104.5°),导致轨道重叠较差(不利,但比杂化更有利)。
我听说过的第三种方法是本特规则(Henry bent),它说在sp^n杂化轨道中,n不一定是整数。所以孤对电子的轨道有更多的s轨道(因为离原子核更近)而氢的轨道有更多的p轨道(因为离原子核更远)。
也许有一天,如果你有时间和资源,你可以讨论这个话题。
我很想听听你对这件事的看法。
奥地利向你致以最诚挚的问候。
本文的工作表明泡利原理不适用于化学键,并基于海森堡测不准原理提出了一种新的化学键理论模型。
见第88 - 104页。基于三电子键的苯。(泡利不相容原理,海森堡测不准原理和化学键)。http://vixra.org/pdf/1710.0326v1.pdf
泡利不相容原理和化学键。
泡利不相容原理——这是量子力学的基本原理,它断言两个或多个相同的费米子(具有半积分自旋的粒子)不能同时处于相同的量子态。
瑞士理论物理学家沃尔夫冈·泡利(Wolfgang Pauli)在1925年提出了这一原理。在化学中,泡利不相容原理常被认为是禁止倍数为1.5的三电子键的存在,但可以证明,泡利不相容原理并不禁止三电子键的存在。要做到这一点,更详细地分析泡利不相容原理。
根据泡利不相容原理,在由相同费米子组成的系统中,两个(或多个)粒子不可能处于相同的状态[2]。波函数和行列式的相应公式在参考文献中给出(这是费米子系统的标准考虑),但我们将注意力集中在推导上:“……当然,在这个公式中,泡利不相容原理只能应用于弱相互作用粒子的系统,当一个人可以说话(至少近似于单个粒子的状态)”[2]。也就是说,泡利不相容原理只能应用于弱相互作用粒子,当我们可以讨论单个粒子的状态时。
但是,如果我们回想一下,任何经典的化学键都是在两个原子核之间形成的(这是与原子轨道的根本区别),它以某种方式“拉”电子到另一个原子核上,那么假设在化学键的形成过程中,电子不再被视为弱相互作用的粒子是合乎逻辑的。这一假设被前面介绍的化学键作为独立的半虚粒子的概念所证实(粒子“部分”的自然成分不能弱相互作用)。
在“海森堡的不确定性原理和化学键”一章中给出的化学键的表示断然拒绝了关于化学键是弱相互作用电子系统的陈述。相反,从上面的描述可以得出,在化学键中,电子“失去”了自己的个性,“占据”了整个化学键,即化学键中的电子“尽可能多地相互作用”,这直接表明泡利不相容原理对化学键的不适用。此外,动量和坐标的量子力学不确定性实际上严格地表明,在化学键中,电子是一个“最大”强相互作用的粒子系统,整个化学键是一个单独的粒子,其中没有“单个”电子的概念,它的速度、坐标、能量等描述。这根本是不对的。化学键是一个单独的粒子,我们称之为“半虚粒子”,它是由单个电子(强相互作用)组成的复合粒子,并在空间上位于原子核之间。
因此,引入倍数为1.5的三电子键从化学的角度(简单地解释了苯分子的结构、芳香性、有机和无机物的结构等)是合理的,被泡利不相容原理和化学键作为强相互作用粒子系统(实际上是一个独立的半虚粒子)的逻辑假设所证实。因此泡利不相容原理不适用于化学键。
1.泡利W.优步登Zusammenhang在原子结构的复杂结构和光谱的研究中,- Z.物理。, 1925, 31,765 -783。
2.A.S.达维多夫。量子力学。第二版。《科学》出版社。莫斯科,1973年,第334页。
海森堡的测不准原理和化学键。
为了进一步分析化学键,让我们考虑电子的康普顿波长:
λе。= h/(me*c)= 2.4263 * 10^(-12) m
电子的康普顿波长相当于光子的波长,其能量等于电子本身的剩余能量(标准结论如下):
λ= h / (m * v), E = h *γ,E =我* c ^ 2, c =γ*λ,γ= c /λ
E = h*γ, E = h*(c/λ) = me*c^2, λc。= h / c(我*)
其中λ是路易斯·德布罗意波长,me是电子质量,c, γ是光的速度和频率,h是普朗克常数。
更有趣的是考虑在线性维度小于电子康普顿波长的区域中电子会发生什么。根据这一领域的海森堡不确定性,我们在动量上至少有m*c的量子力学不确定性,在能量上至少有me*c^2的量子力学不确定性:
Δp≥me*c和ΔE≥me*c^2
这足以产生虚电子-正电子对。因此,在这样的区域中,电子不再被视为“点物体”,因为它(一个电子)的部分时间处于“电子+对(正电子+电子)”的状态。因此,一个距离小于康普顿长度的电子是一个具有无限个自由度的系统,它的相互作用应该在量子场论的框架内描述。最重要的是,向中间态“电子+对(正电子+电子)”的跃迁是以时间为单位的~ λc.е./c
Δt = λc.。/c = 2.4263 * 10^(-12)/(3*10^8) = 8.1*10^(-20) s
现在,我们将尝试用上述所有的方法,用爱因斯坦的相对论和海森堡的测不准原理来描述化学键。为了做到这一点,让我们做一个假设:假设在玻尔轨道上的电子(氢原子)的波长与电子的康普顿波长相同,但在另一个参照系中,结果是康普顿波长大137倍(由于相对论的影响):
λе。= h/(me*c) = 2.4263 * 10^(-12) m
λb。= h/(me*v)= 2*π*R = 3.31*10^(-10) m
λ/λc。е。= 137
其中R= 0.527 Å,波尔半径。
由于氢原子中的德布罗意波长(根据玻尔)比电子的康普顿波长大137倍,因此可以很合理地假设能量相互作用将弱137倍(光子波长越长,频率越低,因此能量也就越低)。我们注意到1 / 137.036是一个精细结构常数,这个基本物理常数是1916年由德国物理学家Arnold Sommerfeld引入科学的,用于描述N.玻尔原子模型框架内原子光谱的相对论修正。
为了描述化学键,我们使用海森堡测不准原理:
Δx * Δp≥/ 2
考虑到能量相互作用减弱了137倍,海森堡测不准原理可以写成这样:
Δx* Δp≥(口* 137)/2
根据最后一个方程,化学键中电子动量的量子力学不确定性必须至少为me * c,能量中的量子力学不确定性不小于me * c ^ 2,这也应该足以产生虚电子-正电子对。
因此,在化学键领域,在这种情况下,一个电子不能被视为“点物体”,因为它(一个电子)会有一部分时间处于“电子+对(正电子+电子)”的状态,因此它的相互作用应该在量子场论的框架中描述。
这种方法可以解释在多电子化学键(两电子、三电子等)的情况下,如何克服电子之间的排斥力:由于化学键实际上是电子和正电子的“沸腾质量”,虚正电子“帮助”克服电子之间的排斥力。这种方法假设化学键实际上是一个封闭的空间袋(能量意义上的势阱),其中发生了真实电子的“沸腾”,也发生了虚拟正电子和电子的“沸腾”,这个势袋的“体积”实际上是化学键的“体积”,也是电子位置量子力学不确定性的空间度量。
严格地说,有了这样的考虑,电子不再具有一定的能量、动量、坐标,不再是“点粒子”,而是实际上占据了化学键的“整个体积”。可以说,在化学键中,单个电子是去人格化的,失去了它的个体性,实际上它并不存在,而是存在由真实电子、虚拟正电子和电子组成的“沸腾质量”,它们通过波动相互改变。也就是说,化学键实际上是一个单独的粒子,就像前面提到的,一个半虚粒子。此外,这种方法可以扩展到基本粒子的结构,如电子或正电子:在这种情况下,基本粒子是封闭在某个空间袋中的波动真空,这是这些波动的势阱。
特别值得注意的是,在这种考虑下,电子是强相互作用的粒子,因此泡利原理不适用于化学键(详情请参见“泡利原理与化学键”一节),也不禁止相同的多重数为1.5的三电子键的存在。
上面的例子很容易用长度为1 Å的化学键来证明。则布罗意波的波长为(化学键长度为L = 2 * Δx):
λ = 2*π*Δx
海森堡不确定性比的形式为:
Δx * Δp≥(* 137 * 2 * π) / 2
从中我们得到:
L * Δp≥* 137 * 2 * π
其中L是化学键的长度,Δp是给定化学键中每个电子动量的量子力学不确定性。
由此,我们得到了化学键动量不确定度的公式:
Δp≥(π * 137 * 2 * π) / L
在对长度为1 Å进行了必要的计算后,我们得到:
Δp≥(* 137 * 2 * π) / 10 ^ (-10)
Δp≥9.078 * 10 ^ (-22)kg * m / s
也就是说,脉冲的不确定性大于me * c
(me * c = 2.73 * 10 ^ (-22) kg * m / s)
(很明显,电子速度的不确定性将大于光速),这应该是基于我们的假设。
见第88 - 104页。基于三电子键的苯。(泡利不相容原理,海森堡测不准原理和化学键)。http://vixra.org/pdf/1710.0326v1.pdf
苯:以三电子键为基础的苯:
审查。基于三电子键的苯。(泡利不相容原理,海森堡测不准原理和化学键)。http://vixra.org/pdf/1710.0326v1.pdf
1.苯分子以三电子键为基础的结构。
http://vixra.org/pdf/1606.0152v1.pdf
2.实验证实了三电子键的存在及其存在的理论依据。
http://vixra.org/pdf/1606.0151v2.pdf
3.化学键的简短分析。
http://vixra.org/pdf/1606.0149v2.pdf
4.补充了三电子键存在的理论证明。
http://vixra.org/pdf/1606.0150v2.pdf
5.并对四部著作中的三电子键理论作了简要评述。
http://vixra.org/pdf/1607.0022v2.pdf
6.审查。苯以三电子键为基础。http://vixra.org/pdf/1612.0018v5.pdf
7.鲍林共振理论的量子力学方面。
http://vixra.org/pdf/1702.0333v2.pdf
8.从PQS的位置对MO法和VB法进行量子力学分析。
http://vixra.org/pdf/1704.0068v1.pdf
9.审查。基于三电子键的苯。(泡利不相容原理,海森堡测不准原理和化学键)。http://vixra.org/pdf/1710.0326v1.pdf
Bezverkhniy Volodymyr (viXra):http://vixra.org/author/bezverkhniy_volodymyr_dmytrovych