烯醇、烯醇化物
烯醇-催化醇醛的反应、卤化和曼尼希反应
最后更新:2023年5月22日|
烯醇-醇醛缩合反应,卤化,曼尼希反应
在先前的文章中酮烯醇互变现象,我们描述了酮烯醇形式。在本帖里,我们探索的反应性烯醇互变异构体,可以作为亲核试剂在一些重要的反应。
在本帖里,我们将有很多要说酸催化的作用,帮助促进酮烯醇互变现象,以及讨论酸如何加速反应的”的共轭酸是一个更好的亲电试剂”和“的共轭酸是一个更好的离去基团”。
表的内容
1。烯醇的反应
在以前的文章中我们探讨酮烯醇互变现象(看到帖子:Keto-Enol互变现象),描述了烯醇互变异构体是亲核自我的亲电酮互变异构体。在本文中,我们将进入一些更实际的方面,即:什么样的反应烯醇经历吗?
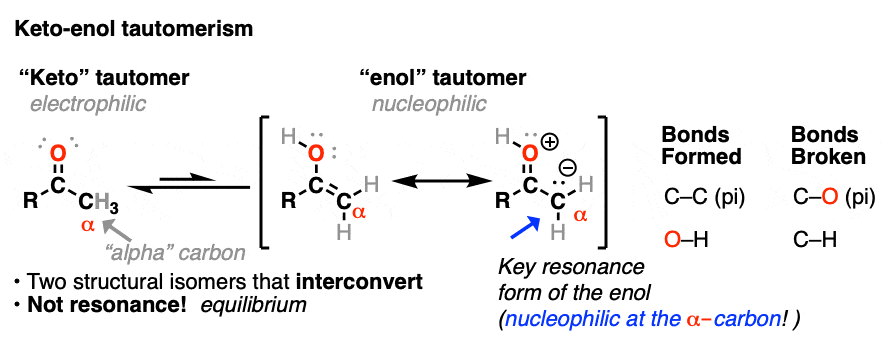
虽然不是那么亲核的烯醇化物(注1),烯醇在几个重要的有机反应中发挥关键作用。
在本文中,我们来看看三个重要的例子:
- 卤化的酮
- 催化醇醛缩合。
- 曼尼希反应(很像醇醛的除外烯醇增加氮π键,而不是切断π键)
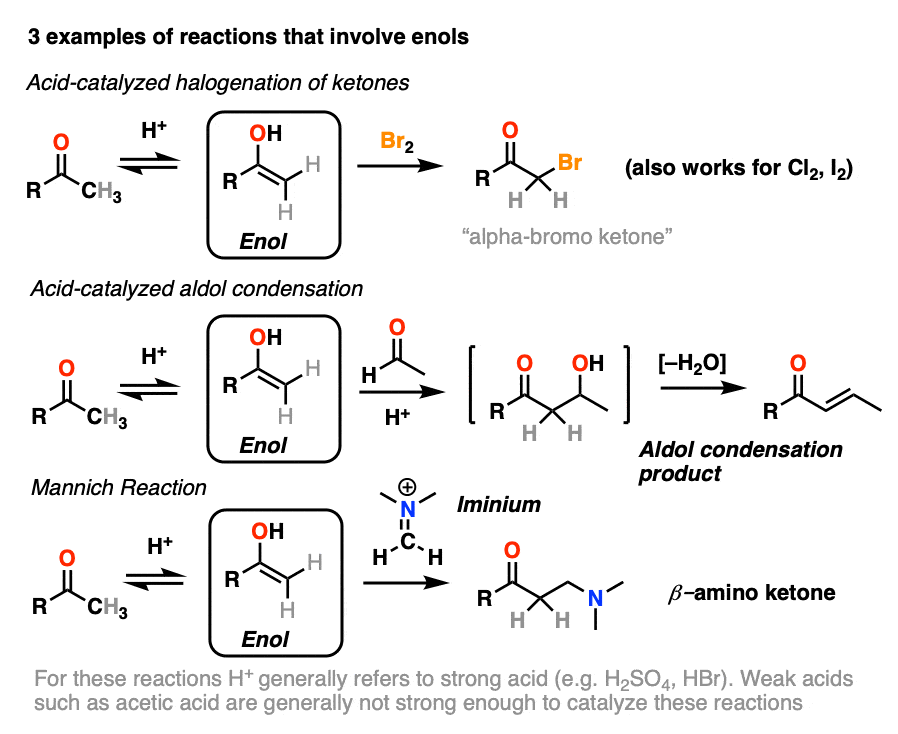
如果你仔细看看上面的三个反应,你会注意到,他们每个人由酸催化。
这是为什么呢?让我们仔细看。
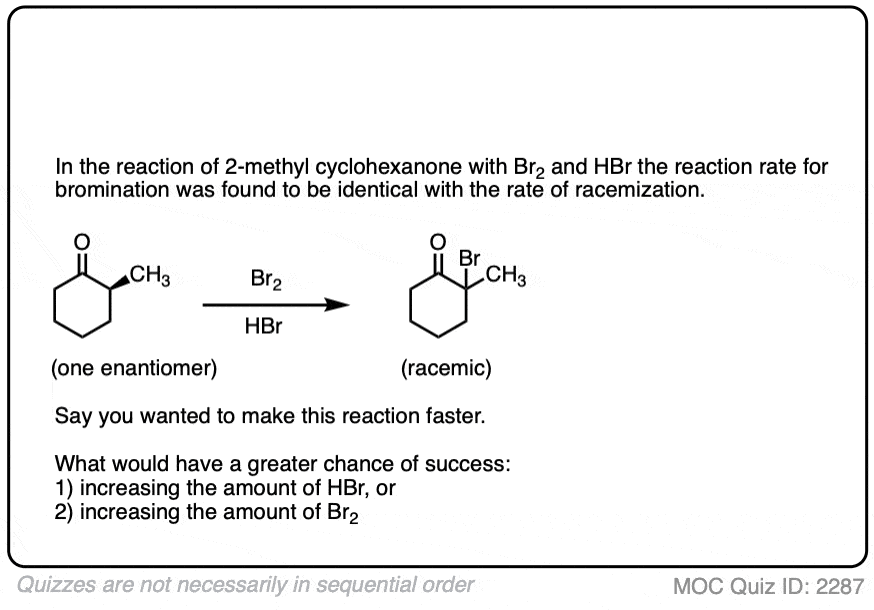
2。在α碳反应:差向异构化作用
让我们创造了条件,反应似乎并没有真正实现。
如果你用光学纯的一个解决方案,旋光性酮像这样的((年代)2-methylcyclohexanone]添加几滴强酸和搅拌过夜,第二天早上你会回来发现它的旋光性完全消失了。(注2- - - - - -醋酸不够强劲,但几滴哈佛商业评论]
它仍然2-methylcyclohexanone,但成为一个平等的混合物(R)和(年代)对映体。一个外消旋混合物换句话说。(见文章:什么是外消旋混合物?)
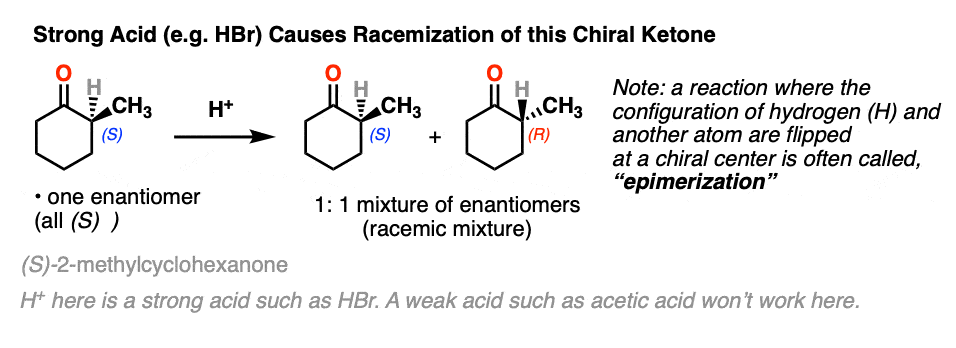
相反,如果你把一个非常类似的光学纯酮(3 -甲基环己酮)和执行相同的序列,光学活动将依然存在不变。
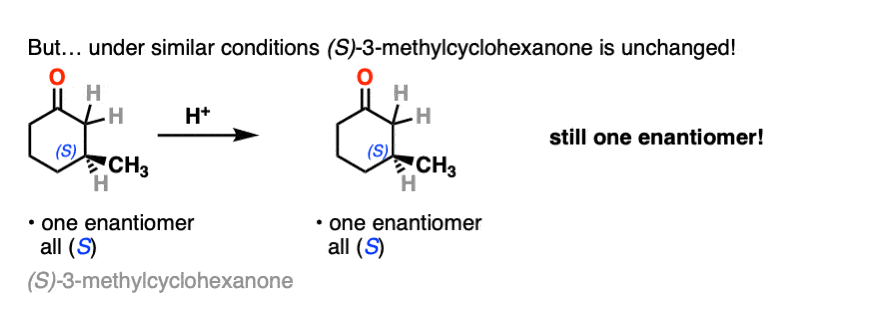
这是怎么回事?
酮都可以进行互变现象给烯醇形式,在c - 2三角形平面(sp2杂化)。然而,烯醇2-methylcyclohexanone,不再有一个手性中心。的烯醇形式是非手性的。
当这个平,非手性的烯醇然后由酸再生2-methylcyclohexanone质子化了的,会这样做吗平等的可能性从顶部和底部的脸上,导致平等(即外消旋)的混合物(右)和(年代)对映体。
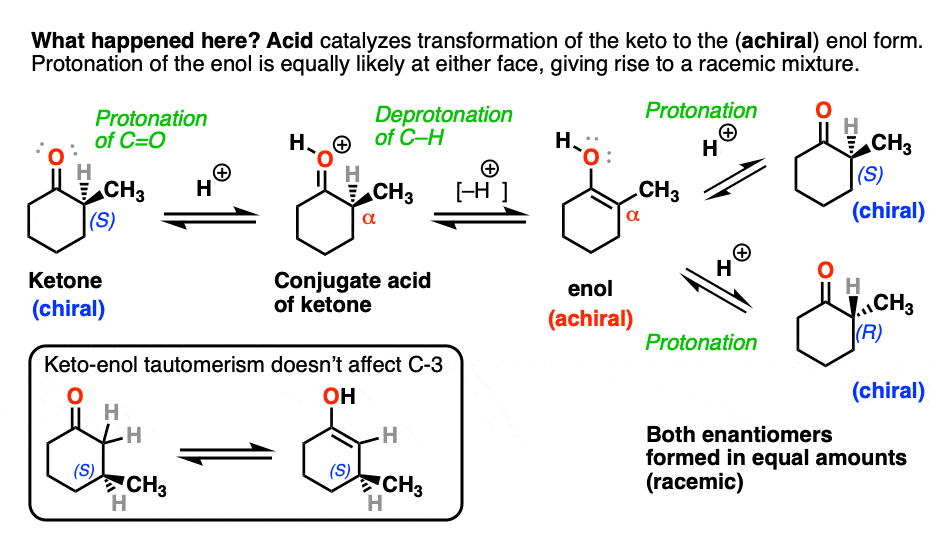
另一方面,(年代)3-methylcyclohexanone保持它的旋光性手性中心以来并没有摧毁时烯醇就形成了。
反应两个取代基对一个手性中心交换(其中一个通常是H)差向异构化作用。
差向异构化作用的α碳酮和醛可以容易发生在酸和碱的存在。碳在链不受影响,因为它是唯一的α碳这是酸性的。
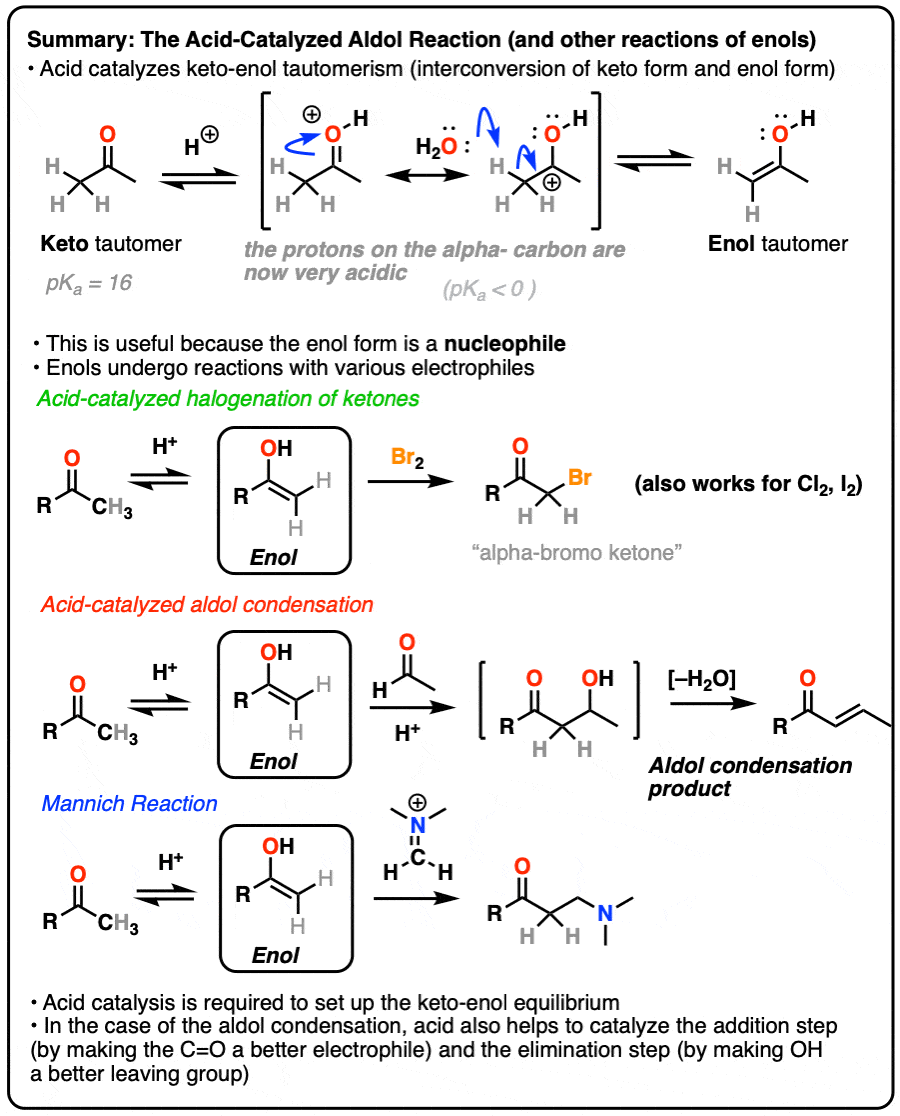
3所示。到底是如何Keto-Enol互变异构催化酸?
同样的实验被执行吗没有酸吗?是的,但它会慢得多。
酸有助于催化酮烯醇互变现象,允许快速之间的平衡酮和烯醇形式。
如何具体地说酸有帮助吗?
如果你走在时间组织1,你会发现反应深埋在你的银行叫做记忆E1的反应。(见文章:E1反应及其机制)
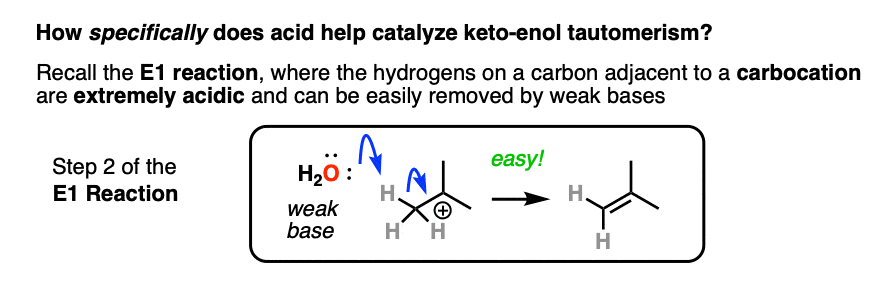
后离去基团叶子,下一步E1是去质子化的碳碳正离子给旁边一个π键。的E1需要只对这一步弱碱(即使水会做!)自离去基团已经离开和碳提供了一个空的p轨道,形成新的π键。(请记住,E2为了得到,需要一个强大的基地离去基团离开-看到帖子:E2机制)。
碳的质子化了的醛或酮非常类似于碳正离子。它有一个明显的共振形成的地方形式电荷碳和氧是中性的。(注3]
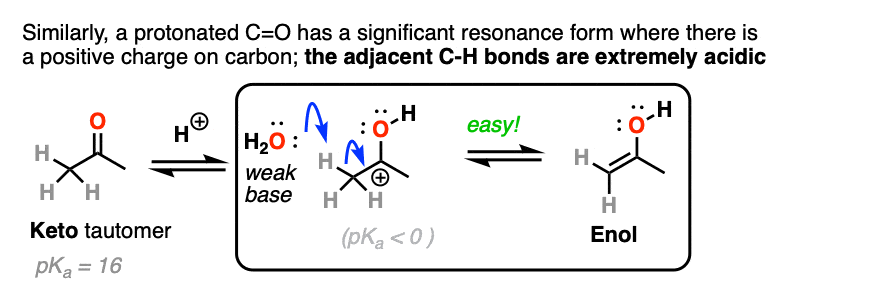
旁边的碳氢键的去质子化羰基(pK一个< 0)因此大大更容易要比在中性的情况醛/酮(pK一个= 16)
TL:博士加酸使它容易deprotonate碳氢键,因此使形成的烯醇容易得多。
酸还帮助的转换烯醇回酮,其中包括(慢)的质子化作用α碳(快速)去质子化的地紧随其后。
的催化转化烯醇酮,徘徊在这里和一个图像会弹出或点击这个链接。
{kind=link}
因为前后反应加速,我们的意思是当我们说酸”有助于建立平衡”。
现在让我们看看一些烯醇的反应。
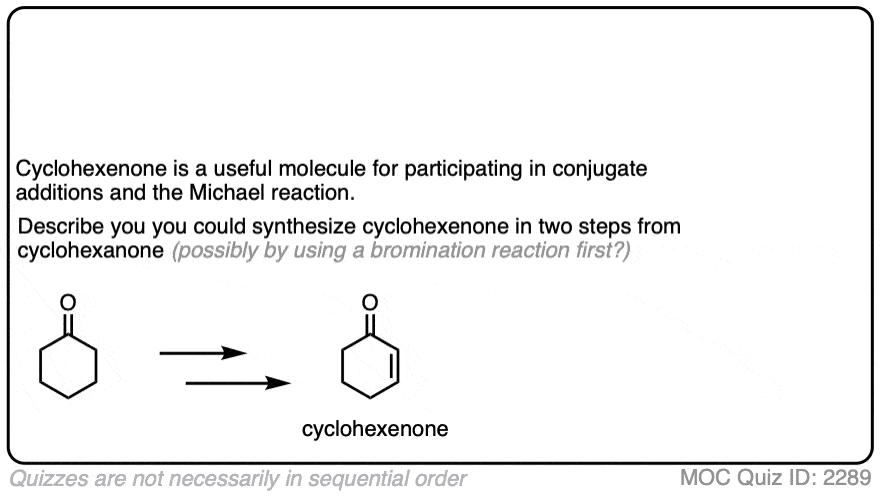
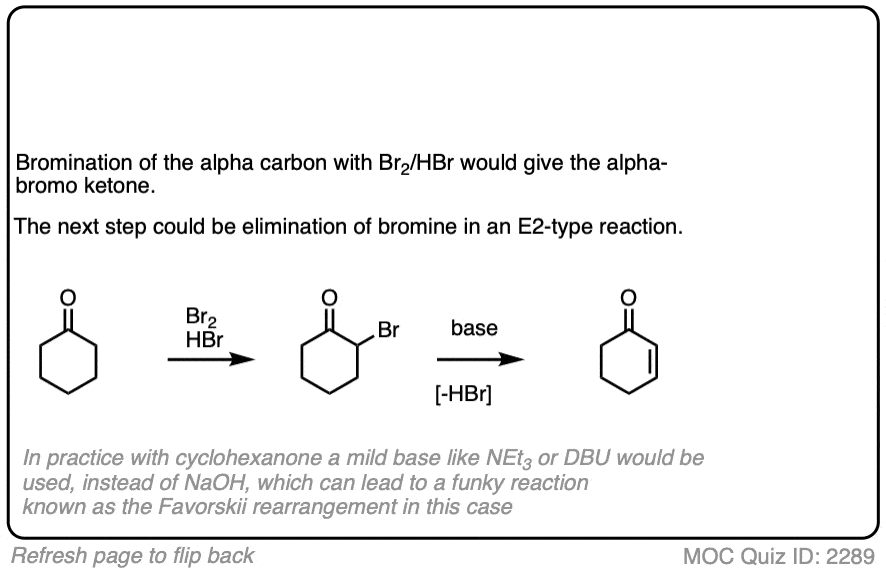
4所示。溴化的酮
烯醇只是负电子烯烃,并将经历许多相同的反应。您可能还记得,烯烃将容易攻击Br等卤素2例如,给二卤化物。(看到帖子:溴化烯烃的)
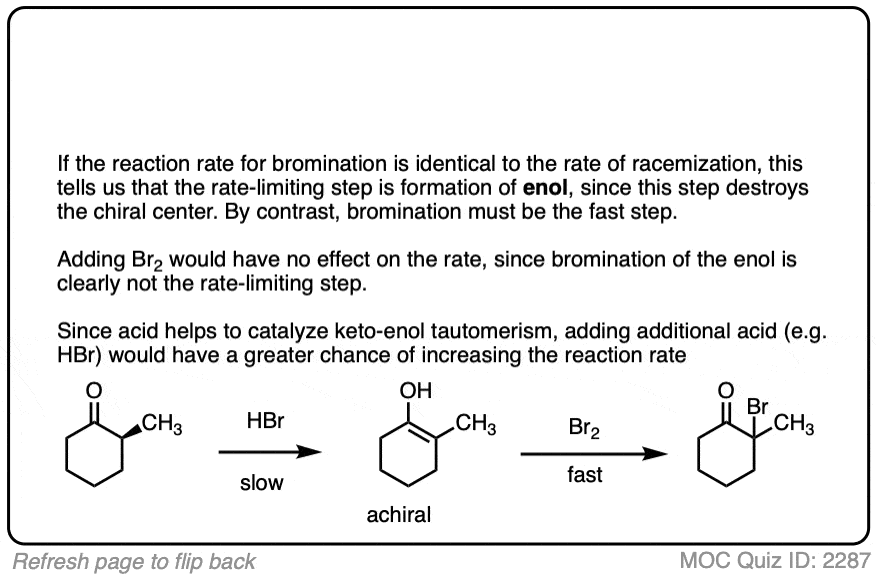
如果你添加强酸酮,混合卤素(氯2、溴2,我2),你得到一个新的alpha-halo酮。
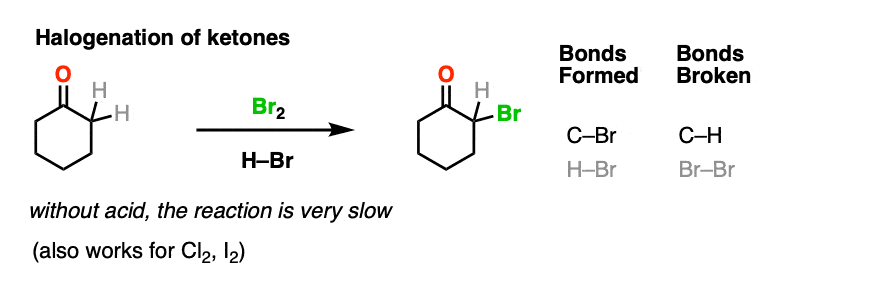
在缺乏酸,反应是“极其缓慢”。(注意4]此外,仔细的测量表明,添加过多的Br2不增加反应速率。
知道了这一点,一个机制,意义是(慢)的转换酮对其烯醇形式,其次是卤化(快)。
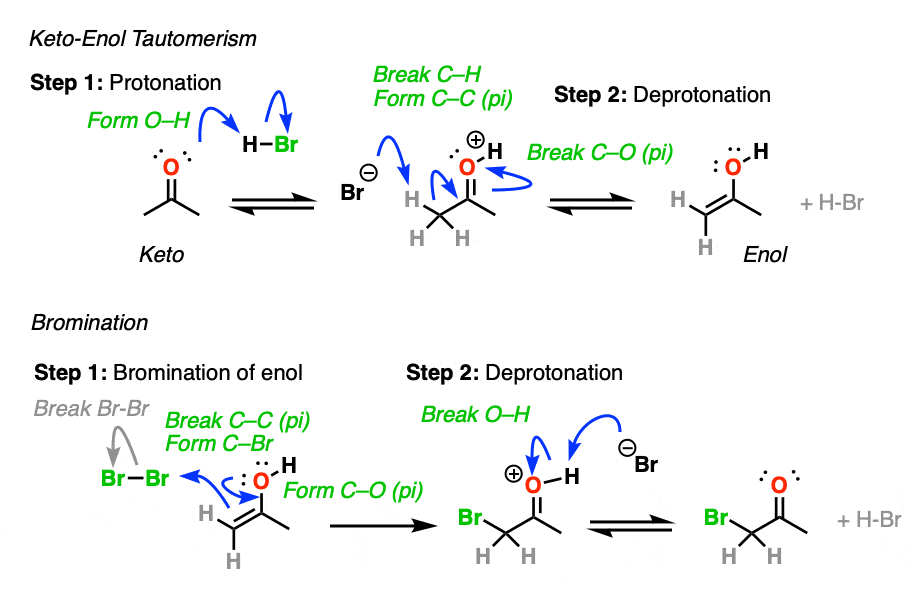
当一个以上的烯醇可以由相同的吗酮,卤素将添加到任何一个烯醇形式是最青睐的平衡(一般最取代——看到的以前的文章在酮烯醇互变现象更多细节)。
 点击翻转
点击翻转
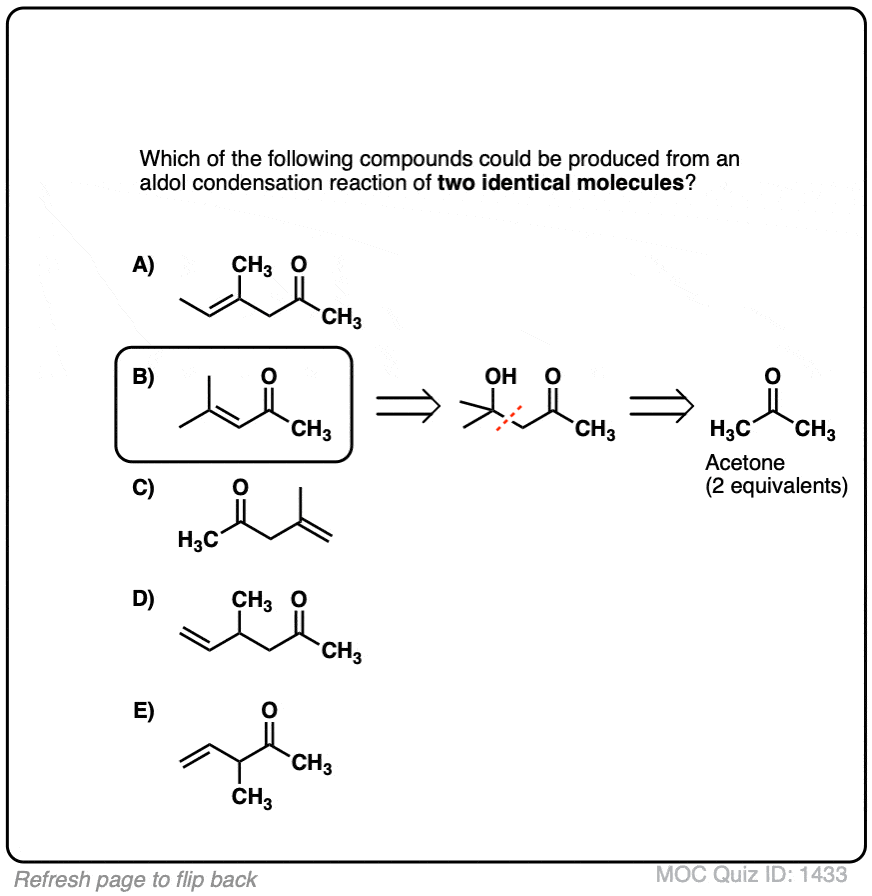
5。催化醇醛反应和酸催化的三个路径
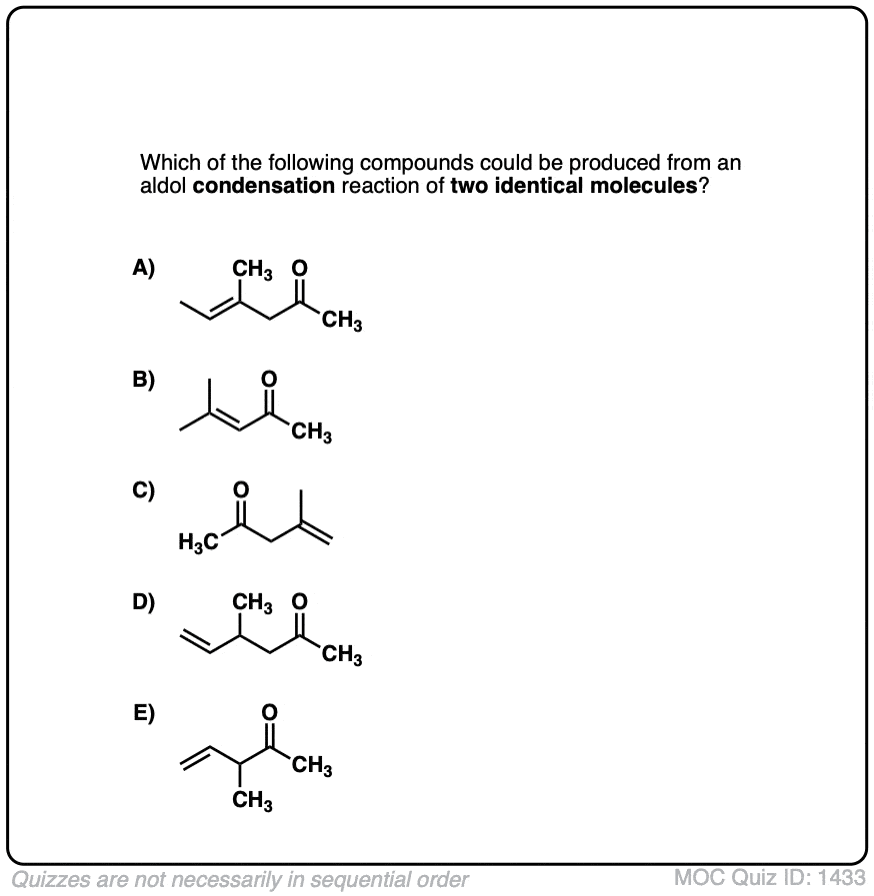
以前我们看到如何共轭碱烯醇(被称为“烯醇化物”)可以添加,醛和酮的反应醇醛反应。(见文章:醇醛加法和冷凝)
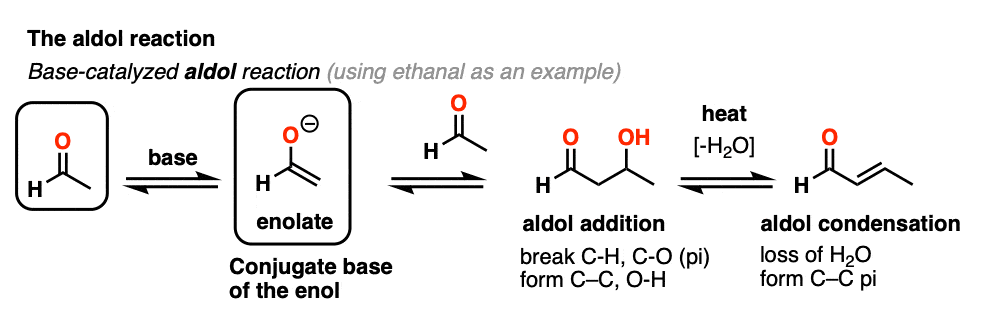
烯醇还可以进行醇醛醛和酮的反应,如果酸催化剂存在帮助搬东西。催化醇醛反应几乎总是导致冷凝产品(H2O),即使没有热量。
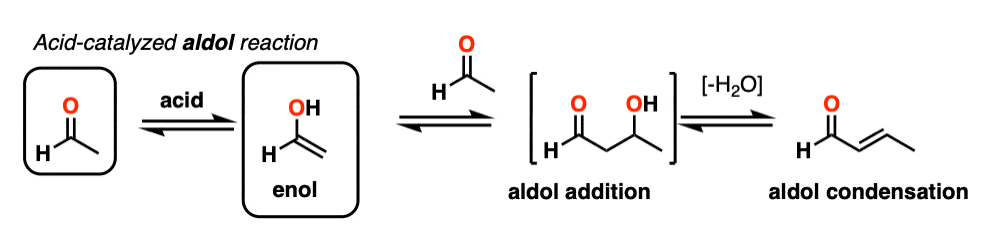
更难控制,催化醇醛往往不如base-catalyzed版本使用。(注5]然而,实际上它是有益的,因为它强调了三个关键方面酸可以促进反应,这大约突显出三个阶段的反应。
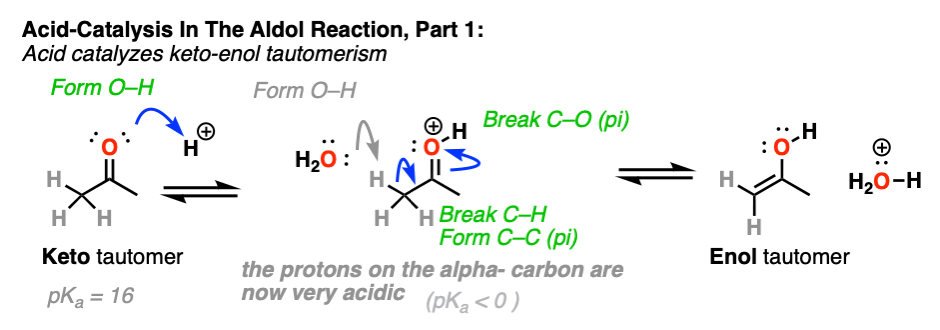
在第一阶段,酸催化酮烯醇互变异构现象,这为我们提供了我们亲核试剂(烯醇),见过之前。
在第二阶段,酸的氧气使质子化醛或酮。由此产生的共轭酸的醛/酮是一个更好的亲电试剂在碳,由于从一个关键贡献共振形式与碳(一个正电荷注3]。
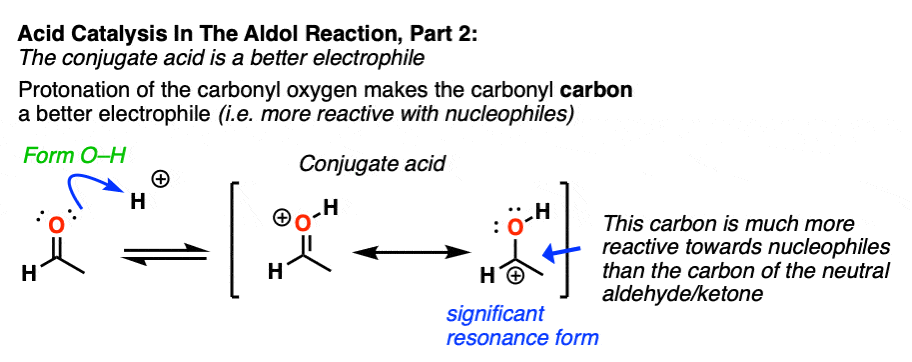
的质子化了的醛/酮然后从亲核的经历之外烯醇,导致醇醛加成去质子化后产品()。
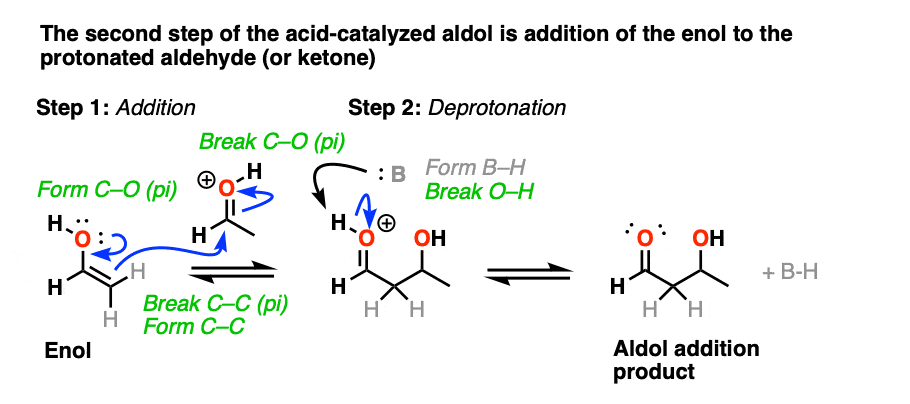
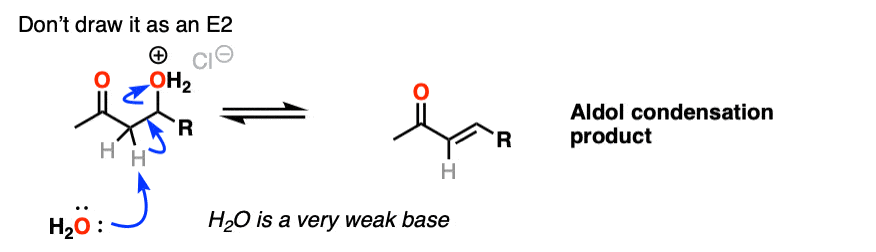
催化醇醛突显出的第三阶段共轭酸是一个更好的离开集团。质子化作用的羟基(OH)之外的产品更好的结果离去基团H2O。
在这个机制的一个版本,水是“挤出”烯醇添加产品的形式。同时形成的切断(pi)和迁移的碳碳(pi)可以被称为“淘汰”或“共轭消除”一步。还有其他方法来显示这种情况,详细的笔记。(注6]
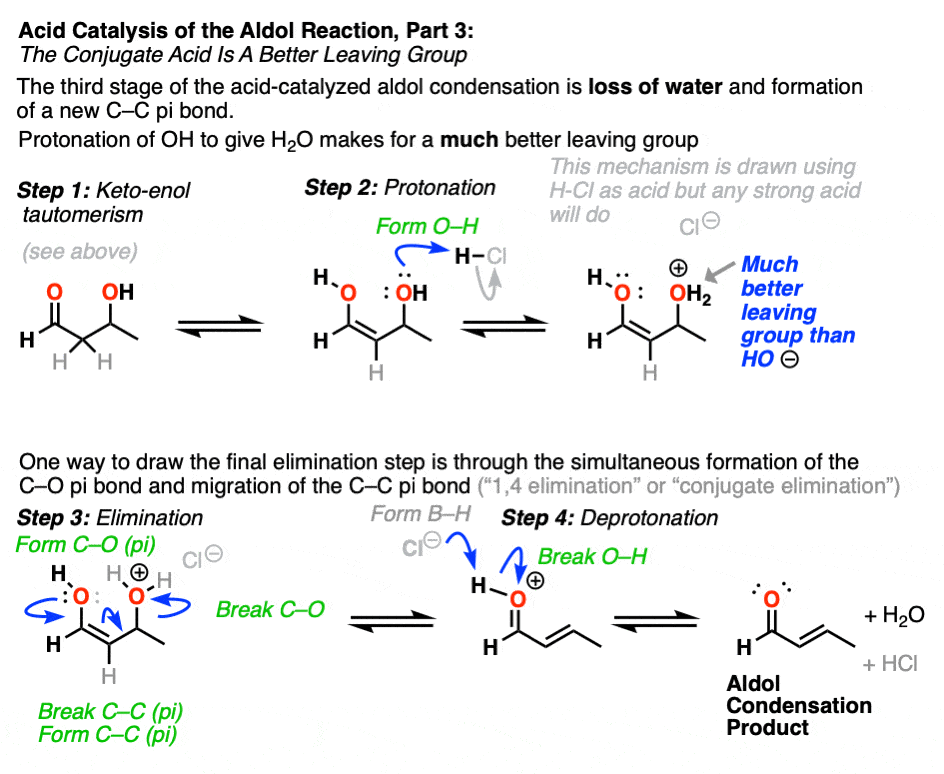
去质子化的氧气然后导致中性醇醛缩合产品。
这里有几个例子,醇醛缩合反应,包括分子内催化醇醛缩合的一个例子。
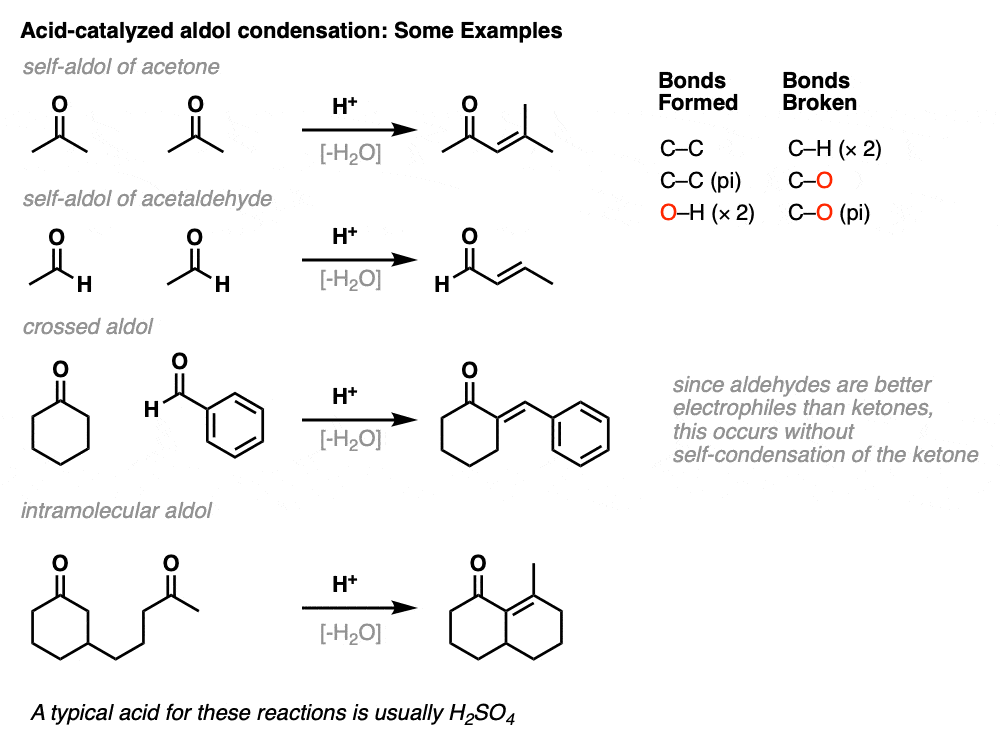
也可以发生分子内催化醇醛的反应。罗宾逊环状结构的一种变体,使用酸,例如。(见文章:罗宾逊的环状结构)
6。曼尼希反应
曼尼希反应是近亲的醇醛反应,除了亲电试剂是一个C = N键而不是C = O。
的亲电试剂在曼尼希通常不是一个中立的亚胺,但带正电iminium离子。
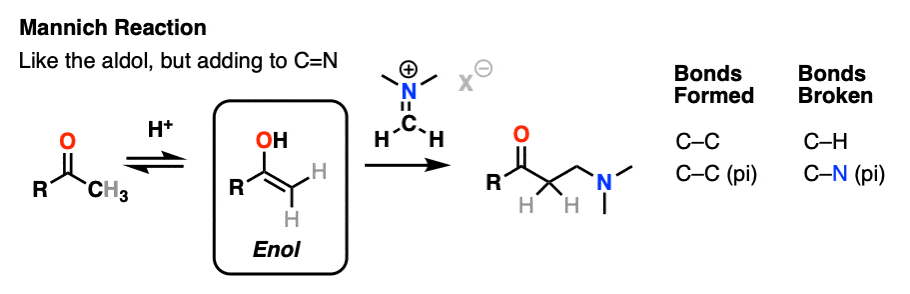
曼尼希反应的第一阶段——你猜对了!-形成的烯醇互变异构体。
在第二阶段,烯醇增加了iminium离子。去质子化然后导致中性产品。
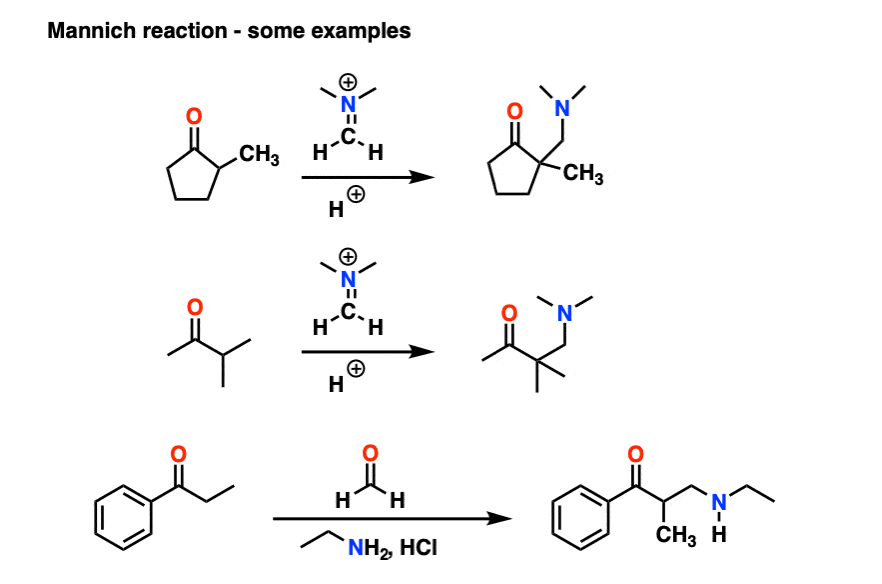
醇醛一样,如果它是在酸性条件下进行,反应将通过取代基更多烯醇。
7所示。结论
我们学到了什么?几件事。
- 烯醇是关键亲核的中间卤化,催化醇醛反应,曼尼希反应。
- 酸催化酮烯醇互变异构现象;的α碳的质子化了的羰基相当多的酸性比中性的吗醛或酮。
- 一个醛或酮一个手性中心α碳将进行差向异构化作用在强酸的存在。
- 对待一个醛或酮与Br2在强酸(哈佛商业评论)将形成一个新的C-Br债券α碳。没有反应发生在缺乏强有力的酸。
- 酸可以促进醇醛缩合。酸有助于促进酮- 1)烯醇互变现象,2)激活羰基对攻击的烯醇亲核试剂,3)有助于消除通过让结果哦这组更好离去基团。
- 的曼尼希反应类似于醇醛反应但需要添加一个烯醇到一个C = N键代替C = O键。它也通过酸催化。
笔记
注1- - - - - -“烯醇不是nucleophlic烯醇化物。”的共轭碱总是更好的亲核试剂比相应的中性的物种。一个量化RO(-)与卢武铉,例如,CH3CH2O(-) 10数量级比CH亲核3CH2哦(见娃的网站这里的来源——你可能要挖]。
没有类似的治疗比较亲核性类似的烯醇化物与烯醇(它们更难研究),但10个数量级可能是一个好猜。
注意2 -”醋酸不够强劲,但几滴哈佛商业评论会”。在一项研究中,人们发现光学纯的外消旋化的速度酮在醋酸/氯仿但极其缓慢的快速在哈佛商业评论的痕迹。(的酮是2-o-carboxybenzylindan-1-one,请参阅j .化学。Soc。1934年773年)。
注意3 -”它有一个明显的共振形成的地方形式电荷在碳和氧气是中性的”。
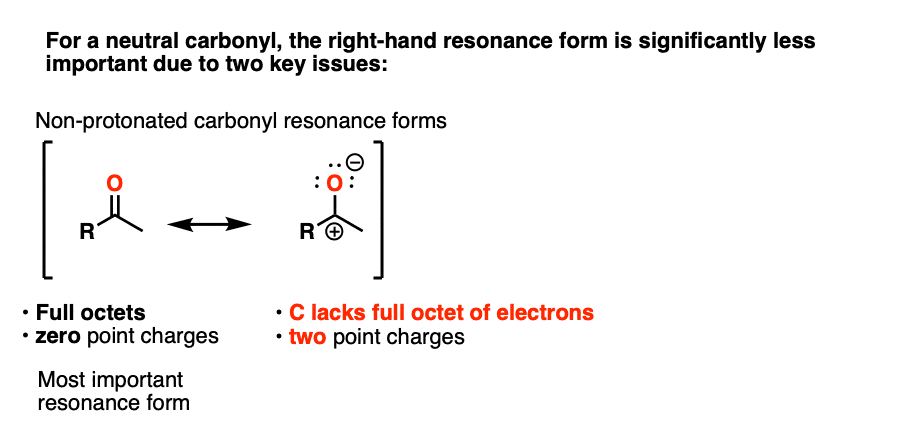
一个直观的方式衡量多少“碳正离子”字符酮是比较的相对贡献carbocation-containing共振形成的整体共振杂化。(直观规则评估的相对重要性共振形式所覆盖,4规则评估共振结构年代)。这里两个关键项目相关点的数量收费(零是首选)和原子的数量与完整的八位字节首选(零)。
的中立酮,共振形成碳正离子两个点费用(C = O和零形式)除了缺乏完整的八隅体C。
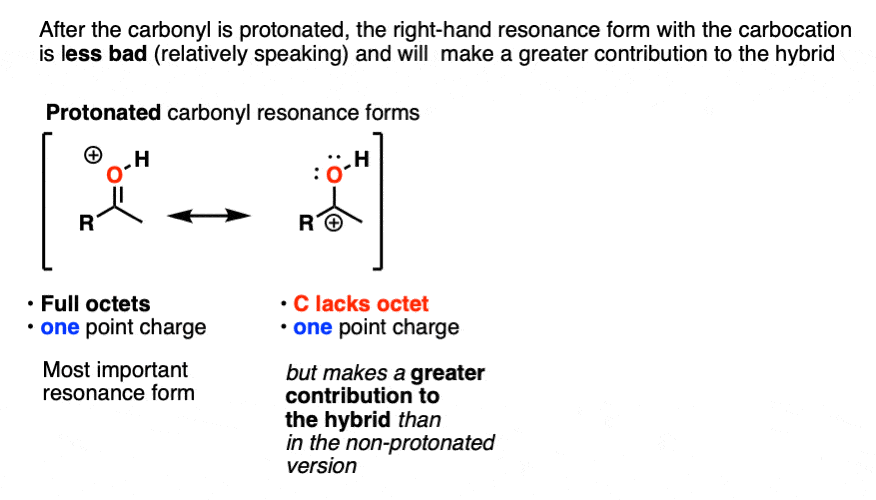
一旦羰基质子化了的,这两个共振形式有相同数量的费用(一个)。因此共振形成碳缺乏一个完整的八隅体(碳正离子)整体不坏,因此,碳正离子会做出更大的贡献共振混合,相对于non-protonated羰基。
量化这种影响的一种方法是检查C = O在红外光谱中延伸,更强的债券以来,波数越高。中性丙酮有一个C = O伸展在1765厘米吗1而质子化了的丙酮C = O伸展在1580厘米1。演示了一个显著削弱切断的π键。
同样的论点可以带正电的原因iminium离子是一个更好的亲电试剂比一个中立的亚胺。
注意4 -”在缺乏酸、溴化极其缓慢”。这是一个直接引用Lapworth (裁判)在他的开创性研究催化溴化。
请注意5 -”的催化醇醛往往不如base-catalyzed版本使用。”的base-promoted醇醛得到更多的使用,因为它是更容易控制烯醇化物你可以通过使用适当的基础形式。在催化醇醛你是哪个人质烯醇热力学形式更稳定。
但是有一个版本的醇醛反应称为Mukaiyama醇醛反应与一个收益烯醇醚随着亲核试剂,拥有强大的路易斯酸(例如TiCl4,抽在空气的存在),协助与醛的反应。
消除通过共轭消除过程是自凝固的决心在这个研究乙醛(见裁判)。
有一些变异这一步是教科书所示,此外,还有变异如何在现实生活中这种情况。
这是有时显示为一种方式E1类型反应水的叶子,有一个后续的去质子化事件。这可以发生;最正确的情况下,将导致共振稳定的碳正离子(如相邻富芳环)。一个著名的例子可以在这里找到。(裁判]
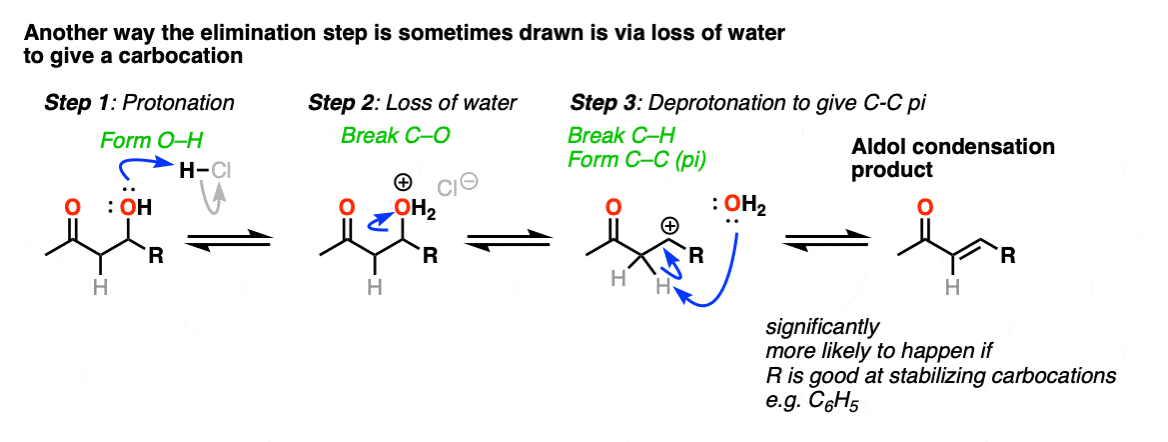
这超越了任何人读这篇文章可能会关心什么,但是也有先例哦组进行替换(可能通过N1)好离去基团,随后被弱碱(或通过E1)。这就是self-aldol凝结的环己酮和盐酸,例如。
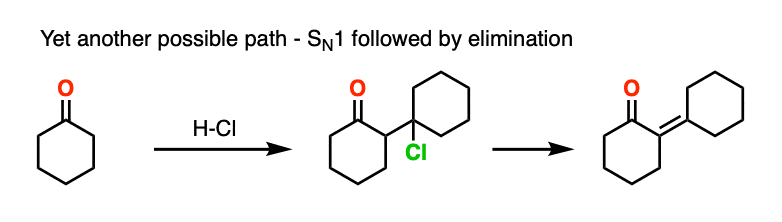
我说的是,你应该能感觉到舒适不画作为一个E2类型的反应。它需要一个anti-periplanar碳氢键和C-OH2债券的安排,我不知道有任何证据表明这个过程取决于立体化学(你看到的相同的方式E2)。
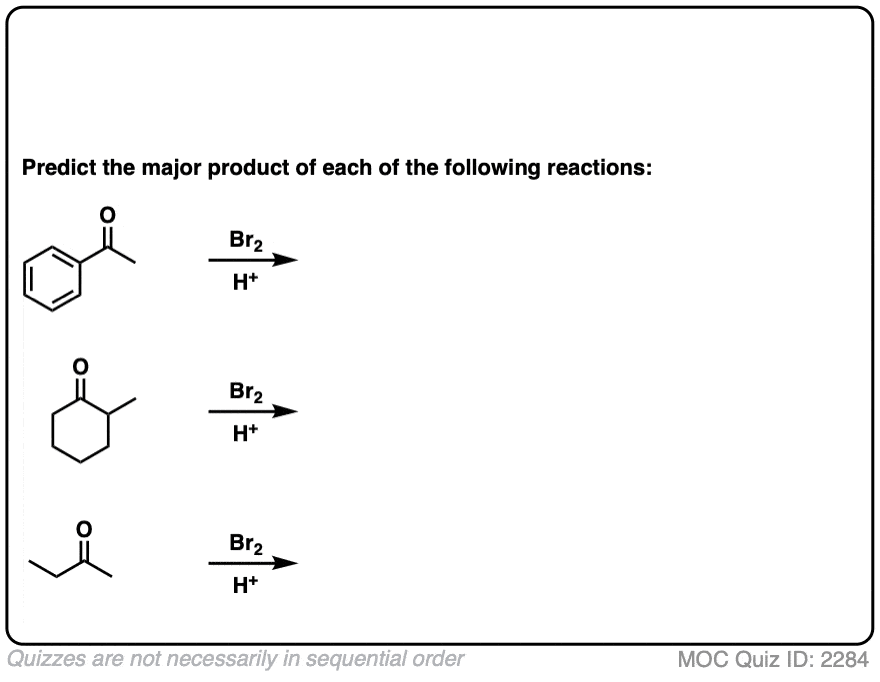
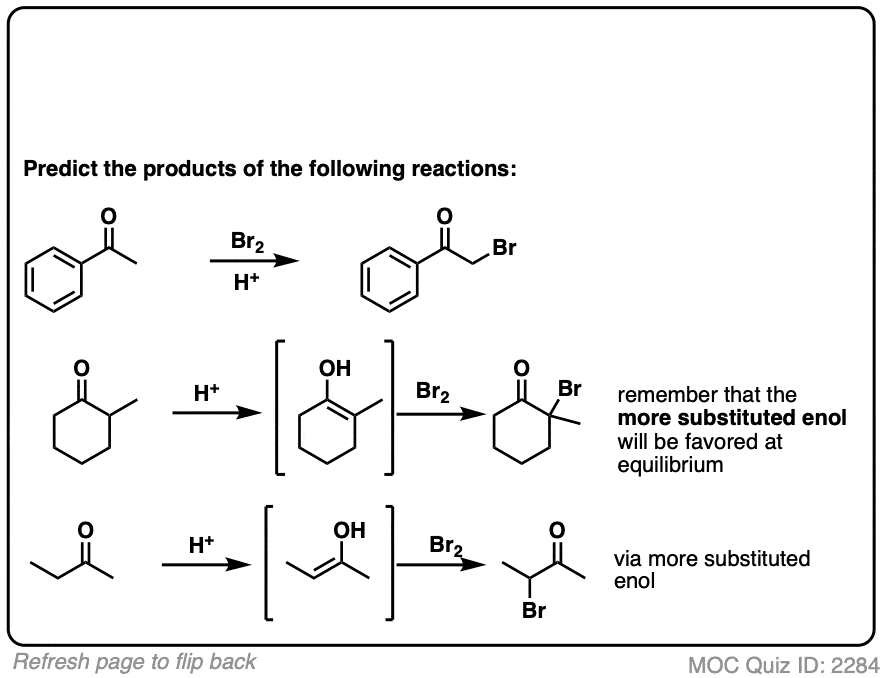
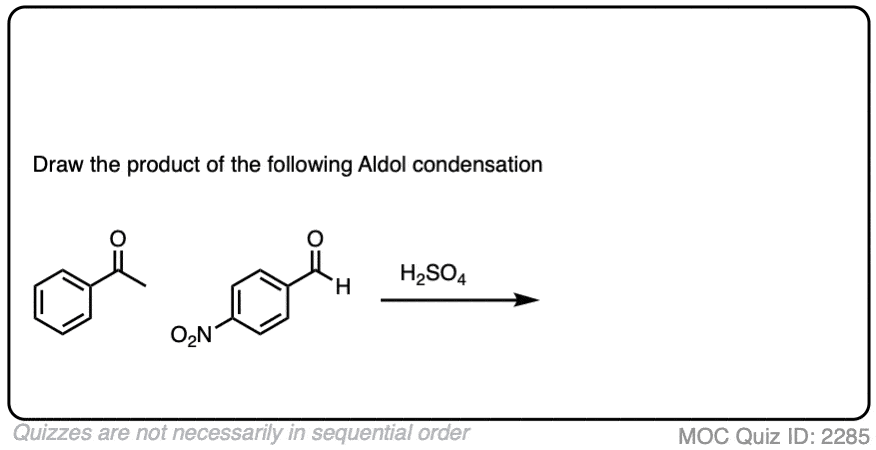
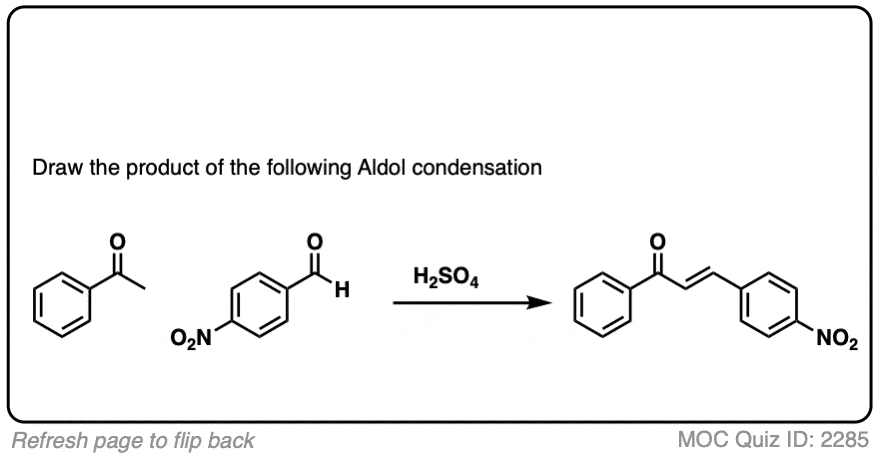
测试你自己!
 点击翻转
点击翻转
 点击翻转
点击翻转

 点击翻转
点击翻转
 点击翻转
点击翻转
 点击翻转
点击翻转
 点击翻转
点击翻转
 点击翻转
点击翻转
(高级)引用和进一步阅读
卷2综合有机合成是有用的对于本文,3月的先进的有机化学,凯莉和桑德博格,在有机化学和英格尔德的结构和机制。
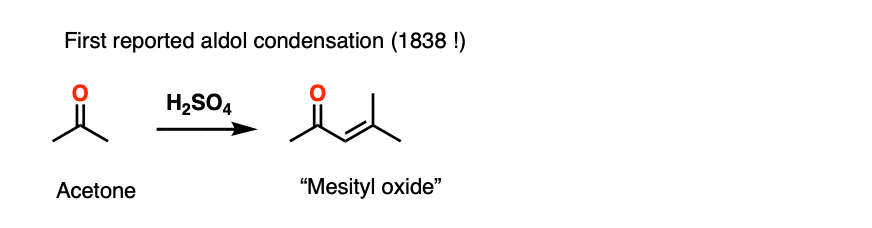
催化醇醛反应已经知道自1838年以来,这使得它的一个最漫长的有机反应。第一个例子的二聚作用丙酮,从j . Prakt。1838年化学,129人。
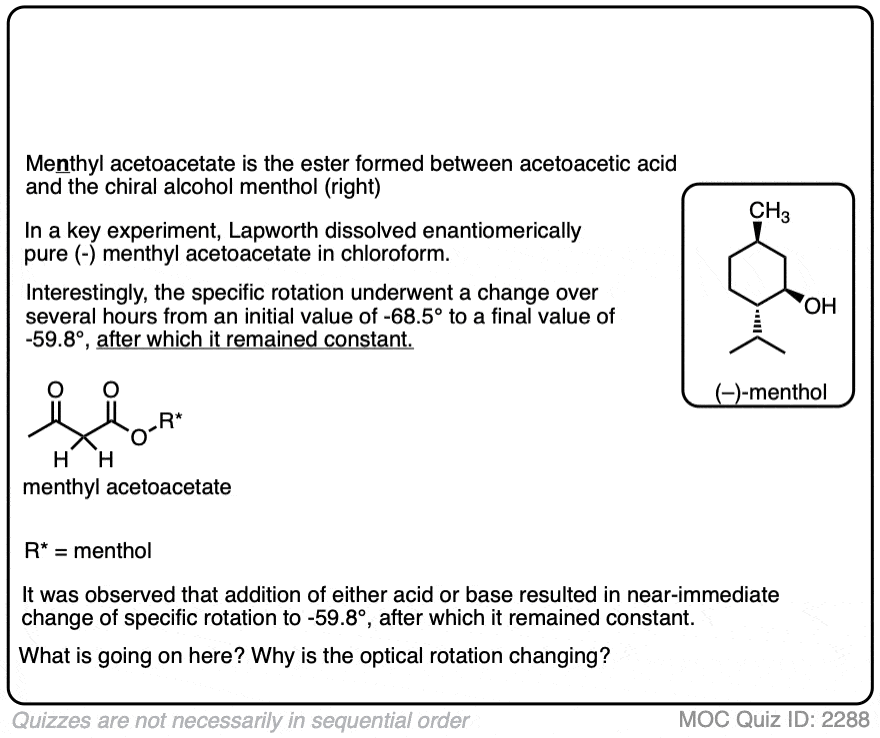
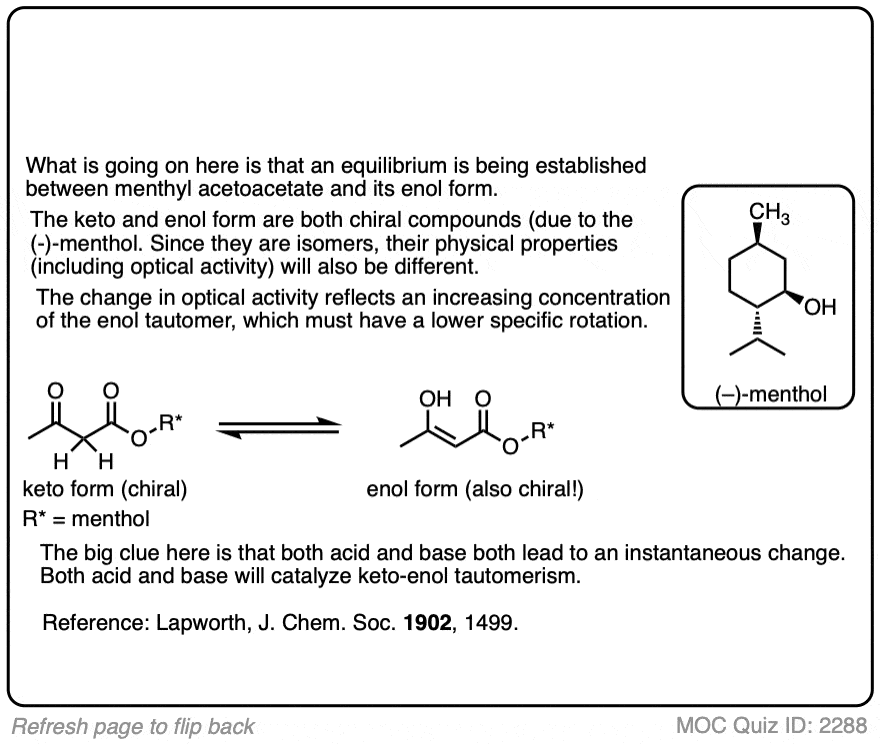
1。光学活性的酯类B-Ketonic B-Aldehydic酸。第二部分。薄荷基乙酰乙酸盐
亚瑟Lapworth和a·c·奥斯本损害。
j .化学。Soc。,反式。1903年,83年,1114 - 1129
DOI:10.1039 / CT9038301114
一个非常有益的纸和最早的例子包含明确的证据在加速酮酸的作用烯醇互变现象。作者准备我n肌酸(不甲基)酯乙酰乙酸((+)薄荷醇是一种旋光性酒精现成的从自然资源),测量其旋光性。他们发现我n肌酸酯经历了旋光改变在某种程度上依赖于溶剂,旋光改变的速率是高度依赖于酸。这说明建立一个酮和之间的平衡烯醇形式。
2。操作包含卤素的化合物羰基集团
亚瑟Lapworth
j .化学。Soc。反式。,1904年85年,30-42
DOI:10.1039 / CT9048500030
卤化的开创性研究羰基化合物。
“溴化丙酮条件下保持最好被看作是一个缓慢的结果,可逆变化的影响丙酮的氢离子,紧随其后的是一个几乎瞬时的溴化产品,这不是明显可逆的变化。这中间产品可能的烯醇的形式酮,因为它已经表明,在许多情况下,快速实现的互变异构的形式之间的平衡羰基化合物是由酸。”
3所示。Ueber死凝结冯Aethylmethylketon麻省理工学院Benzaldehyd(甲乙的缩合反应酮与苯甲醛)
c·哈瑞斯·g·汉斯·穆勒
的误码率。1902年35 (1),966 - 971
DOI:10.1002 / cber.190203501156
乙基甲基的凝结酮与苯甲醛在酸性条件下结果取代基更多的醇醛缩合在高收益产品!
4所示。旋光性与互变异构的改变。第四部分,比较的外消旋化和溴化酮
c·k·英格尔德和c·l·威尔逊
j .化学。Soc。1934年,773年。
DOI:10.1039 / JR9340000773
溴化率基本上是一样的外消旋化,符合烯醇形成病原反应步骤。“在氯仿和冰醋酸,外消旋化d- - - - - -酮非常缓慢而变得非常快速的溴化氢的痕迹”。
5。羰基反应。VI。证据备用机制β-Hydroxy酮的脱水1
唐纳德·s·诺伊斯和威尔默·l·里德
美国化学学会杂志》上1958年80年(20),5539 - 5542
DOI:10.1021 / ja01553a056
本文研究了催化甲基乙基醇醛反应酮和各种取代苯甲醛。金块的关键是,最后脱水步骤提出了通过发生烯醇形式的添加产品当芳环electron-neutral (Ph)或电子(不差2)集团帕拉位置,但是通过电离(即。苄形成碳正离子)富(甲基)集团帕拉的位置。说原因是只有一个3折的利率变化消除一步从p-methyoxyl 3-nitro,与什么人会不一致是否涉及苄阳碳离子。
值得注意的是,唐纳德·诺伊斯的哥哥是物理学家罗伯特·诺伊斯英特尔的创始人之一。
6。羰基反应。八世。催化苯甲醛的缩合反应的动力学和p-Nitrobenzaldehyde甲乙酮。一些观察ρ-σ相关性1
唐纳德·s·诺伊斯和劳埃德·r·斯奈德
美国化学学会杂志》上1959年81年(3),620 - 624
DOI:10.1021 / ja01512a029
一系列的第8部分论文从1958 - 59诺伊斯实验室研究催化醛醇甲乙之间的反应酮和取代苯甲醛。
7所示。催化烯醇化作用和乙醛的醇醛缩合
林恩·m·Baigrie罗宾·a·考克斯Henryka Slebocka-Tilk,对于米甲,托马斯Tidwell t
美国化学学会杂志》上1985年107年(12),3640 - 3645
DOI:10.1021 / ja00298a039
广泛的物理有机化学研究催化醇醛缩合的简单醛乙醛,乙醛)。病原的一步是添加的烯醇质子化了的乙醛。值得注意的是他们的工作也符合消除水的发生烯醇添加产品的形式。
两个其他有用片段:“乙醛完全质子化了的它共轭酸在H2所以4比64%。“酮的烯醇化作用众所周知,涉及两个水分子的表演为基础”。
8。催化罗宾逊Annelations
克莱顿·h·和约翰·e·埃利斯约翰·e·McMurry安东尼•Coppolino
四面体信件,12 (52),1971年,4995 - 4996
DOI:10.1016 / s0040 - 4039 (01) 97609 - 9。
本研究包含几个催化罗宾逊环状结构的例子。
9。曼尼希反应2-Methylcyclopentanone和2-Methylcyclo己酮1
赫伯特·o .和Barry m . Trost房子
《有机化学》杂志上1964年29日(6),1339 - 1341
DOI:10.1021 / jo01029a016
几个有用的曼尼希反应的例子表明,他们更倾向于通过替换烯醇。
请仔细检查“酸催化的醇醛反应,第1部分:酸催化keto-enol互变现象”反应计划。中心结构(质子化了的丙酮)是一个10电子氧原子卸货和特性。更换一个电子对与一个正电荷的氧原子将解决这个问题。
固定!谢谢你的地方!
詹姆斯,
我非常喜欢这篇文章!谢谢你的工作。
2012年Clayden书(pg 616),和Karty书(pg 912)酸催化脱水声称作为一个E1,羰基,而不是烯醇。
我的感觉是,这种机制包括E1,但从烯醇。事实上,在这种情况下,当水叶,形成碳正离子的好处三共振贡献者:β碳上的正电荷,另一个与羰基碳上的正电荷(双键发生了变化),加上oxycarbenium离子。因此,碳正离子有许多理由一个稳定的中间,如果你正式从oxycarbenium删除H +离子最终产品。
谢谢你写!
诺伊斯研究:https://pubs.acs.org/doi/10.1021/ja01553a056
和得出结论,而E1可以发生,特别是在一个相对稳定的碳正离子,可以形成(例如p-MeOC6CH4)。消除一般通过烯醇。这也是自凝固的乙醛。看到的:https://pubs.acs.org/doi/10.1021/ja01553a056
亲爱的先生,我是一个研究学者,我有机合成领域,我做与酸催化剂,醛酮反应我用2乙酰香豆酮和2,氨基5氯苯甲醛,但我预计产品不来,但产品是2,氨基酸,5氯accetophenone,单晶x射线衍射证实了结构,但是它是如何形成的机理机制请帮我想尝试找到任何建议,
我将仔细检查你的起始原料。
2-butanone和2之间交叉醇醛反应,2-dimethylbutanal, 2-butanone负碳离子/亲核试剂考虑2,2-dimethylbutanal缺乏酸性氢。但没有技术两种不同的亲核试剂,2-butanone可能形式,和不会导致两个独立的产品吗?我明白更多2-butanone酸性氢在碳3,但2-butanone酸性的氢碳1 ?它不能成为亲核试剂,或仅仅是不可能,我们不考虑吗?
先生,为什么不氧烯醇化物离子攻击羰基碳?我读过,羰基碳是亲电试剂和oxyanion是亲核试剂,为什么不会发生呢?
它可能发生在某种程度上,但产品是半缩醛,和半缩醛容易恢复到起始原料。这通常是一个死胡同。
教授,缓慢/醇醛反应的速率决定步骤吗?
你能请尽快回复吗? ?…。急事…
谢谢
在催化醇醛,缓慢的一步是攻击的烯醇醛/酮。但可能比你想的更多信息,我建议:https://pubs.acs.org/doi/10.1021/ja01553a056
我相信一个基绑架α氢进行烯醇化作用是必要的。是这样的不是这样的吗?因为在第二个途径你表明,引入酸烯醇化作用速率增加。
是的,keto-enol互变现象可以通过使用辅助酸或基地。
为什么大多数醇醛缩合反应发生在基地的存在
亲爱的先生,
我是研究员,我的研究领域是有机化学。本网站提供了出色的有机反应和机理的信息。我非常深刻的印象,愿提及很多的感谢。通过这个网站,我得到预期的信息和知识。
感谢你,
你的真诚,
uttam帕蒂尔
谢谢你!
1、淘汰机制是新的我,似乎是一个好主意。但是,通常1,2-elimination机制并不是那么糟糕。当然水作为基础不是很好,如果不是用作溶剂(3月(Ed page1349 6日)说,水作为基地甚至在浓硫酸)。这里+是一个很好的离去基团和水(或卢武铉当使用酒精)接受质子形成氧鎓离子。这可能被视为一个E1消除而不是E2消除。其实我发现两种机制的讨论在英格尔德(page1004 - 1005)。有些人认为Cl -离子为基础代替水来捕获质子形成盐酸。但这是不太可能在hydroxylic溶剂。我不知道哪一个才是最可能的机制。不幸的是,今天没有化学家将做实验来阐明机制,因为这样的项目将不会吸引资金。
如果是直E1可以期望看到重组,特别是主要含酒精饮品的地方。自消除水的基质是肤浅的,发生机制烯醇化作用首先其次是捐赠的一对电子的烯醇对我来说似乎更合理,而不是一个E1通道类型,但根据替换多个机制可能是操作模式。第三烯丙基的醇,例如,这可能是主要的机制。
谢谢你花时间置评,也从3月和英格尔德添加注释。