首页/关于羰基的常见错误:羧酸是酸!
有机化学学习小贴士
关于羰基的常见错误:羧酸是酸!
最后更新:2022年10月31日|
羧酸也是酸。
我知道这是显而易见的。但几乎可以肯定的是,第一次参加Org 2课程的学生偶尔会忘记这一点。
以下是我经常看到的两个常见错误。
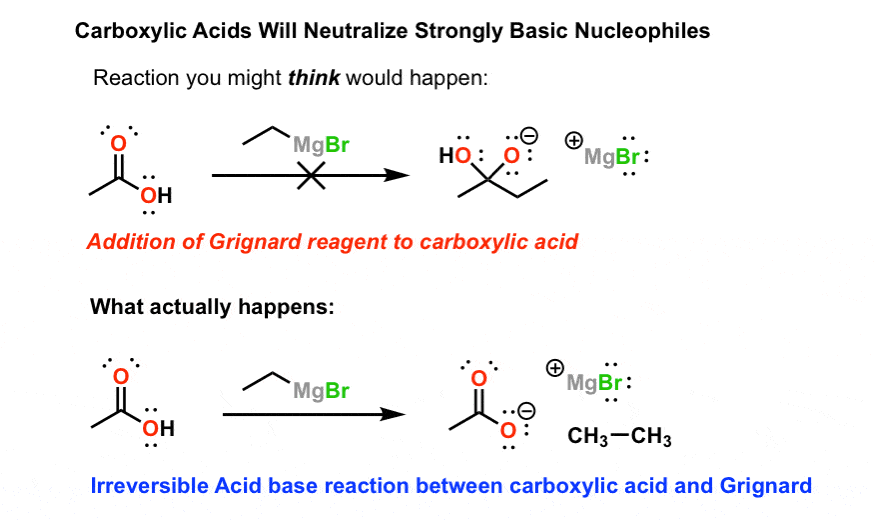
1)格氏试剂与羧酸的反应。
格利雅试剂(具有一般结构RMgBr)是很好的亲核试剂。它们会加入酮,醛,酯类(两次),酸卤化物(两次),环氧化物,以及其他一些羰基包含的化合物。
他们没有。
这是因为羧酸是酸,而格氏试剂是强碱。所以不是加到羰基碳,格氏键首先被质子化。结果是共轭碱的羧酸(一个羧酸盐)反应迟钝,无法进一步反应。
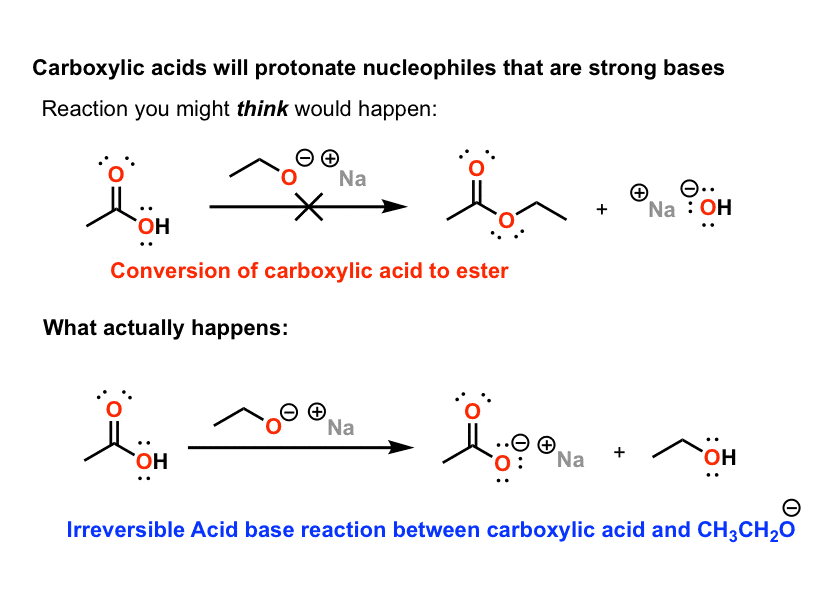
2)与醇氧化合物(脱质子醇)反应
羧酸等衍生品酯类,酐,酸卤化物与HO-和RO-等亲核试剂反应良好。这种模式很快就为人所熟知:1、2之外,然后是1、2取消.看到这种模式,学生们会产生一种错误的安全感,认为羧酸也会这样反应。
它们不会,原因和格氏试剂一样。羧酸也是酸.它们使强碱(如醇氧化合物)质子化,并留下羧酸盐,同样是无反应性的。
重复第三次似乎很愚蠢,但这种情况“一直都在发生”。你可能认为你不会这么做。很有可能,在某些时候,你会。这是一个很容易犯的错误。所以让我们最后再说一遍:
羧酸……酸!
- - -
注意:这是一个很好的经验法则,假设酸碱反应会发生得比亲核攻击的反应快,比如加成反应金属羰基合物.如果你感兴趣,有一个名字可以用来形容:最小运动原理.这就是为什么我们看到格氏试剂质子化在它有时间加入到羰基碳。
01键合、结构和共振
- 我们怎么知道甲烷是四面体?
- 杂化轨道和杂化
- 如何确定杂交:一条捷径
- 轨道杂化和化学键强度
- Sigma键有六种:Pi键有一种
- 关键技能:如何计算形式电荷
- 部分电荷提供了电子流动的线索
- 四种分子间力及其如何影响沸点
- 3种影响沸点的趋势
- 如何利用电负性来确定电子密度(以及为什么不相信形式电荷)
- 共振简介
- 如何使用曲线箭头交换共振形式
- 计算共振形式(1)-最小电荷规则
- 如何利用电负性找到最佳共振结构
- 评价带负电荷的共振结构
- 评价带正电荷的共振结构
- 探索共鸣:派捐赠
- 探索共振:pi受体
- 总结:评价共振结构
- 绘制共振结构:要避免的3个常见错误
- 如何应用电负性和共振来理解反应性
- 键杂化实践
- 结构和结合练习测验
- 共振结构实践
02酸碱反应
03烷烃和命名法
04构象和环烷烃
05有机反应入门
- 学习一种新反应时最重要的问题
- 组织中的4类主要反应
- 学习新的反应:电子如何移动?
- 电子如何(为什么)流动
- 学习一个新的反应时要问的第三个最重要的问题
- 有机化学中稳定负电荷的7个因素
- 有机化学中稳定正电荷的7个因素
- 常见错误:形式上的指控会产生误导
- 亲核试剂和亲电试剂
- 曲线箭头(用于反应)
- 弯曲箭头(2):最初的反面和最后的正面
- 亲核性与碱性
- 三类亲核试剂
- 什么是好的亲核试剂?
- 什么是好的离去基?
- 3个稳定碳正离子的因素
- 碳正离子不稳定的三个因素
- 什么是过渡态?
- 哈蒙德的假设
- 格罗斯曼的统治
- 先画丑的版本
- 学习有机化学反应:核对表(PDF)
- 加成反应简介
- 消除反应简介
- 自由基取代反应导论
- 氧化裂解反应导论
06自由基反应
07立体化学和手性
08置换反应
09消除反应
11SN1 SN2 / E1、E2的决定
12烯烃的反应
- 烯烃的E、Z表示法(+ Cis/Trans)
- 烯烃的稳定性
- 加成反应:消去的反义词
- 选择性vs.特异性
- 烯烃加成反应的区域选择性
- 烯烃加成反应的立体选择性:Syn与Anti加成
- 烯烃中盐酸的马氏加成法
- 烯烃加氢卤化机理及其对马氏规则的解释
- 箭推和烯烃加成反应
- 添加模式#1:“碳正离子途径”
- 烯烃加成反应中的重排
- 烯烃的溴化
- 烯烃的溴化反应机理
- 烯加成模式#2:“三元环”途径
- 硼氢化反应——烯烃的氧化
- 烯烃的硼氢化氧化机理
- 烯烃添加模式#3:“协同”途径
- 溴离子的形成:一个(次要)推箭困境
- 第四种烯烃加成模式-自由基加成
- 烯烃反应:臭氧分解
- 概述:烯烃反应机理的三个关键家族
- 钯对碳(Pd/C)催化加氢
- OsO4(四氧化锇)用于烯烃的二羟基化
- 间氯过氧苯甲酸
- 合成(4)-烯烃反应图,包括卤代烷基反应
- 烯烃反应练习题
13炔的反应
14醇,环氧化合物和醚
- 醇。命名法和性质
- 酒精可以充当酸或碱(以及为什么它很重要)
- 醇类-酸性和碱性
- 威廉姆森醚合成
- 威廉姆森醚合成:规划
- 烯烃、叔烷基卤化物和烷氧汞的醚
- 醇通过酸催化合成醚
- 醚与酸的裂解
- 环氧化物-醚家族中的异类
- 用酸打开环氧化物
- 环氧环开口与基地
- 从醇中制取烷基卤化物
- Tosylates和Mesylates
- PBr3和SOCl2
- 醇的消除反应
- POCl3消除醇生成烯烃的研究
- 酒精氧化:“强”和“弱”氧化剂
- 揭开酒精氧化机制的神秘面纱
- 醇和醚的分子内反应
- 酒精保护团体
- 硫醇和硫醚
- 计算碳的氧化态
- 有机化学中的氧化与还原“,
- 氧化梯子
- 醇制卤代烃的SOCl2机理:SN2 vs SNi
- 酒精反应路线图(PDF)
- 酒精反应练习题
- 环氧反应测验
- 氧化还原练习测验
15有机金属化合物
16光谱学
17Dienes和MO理论
- 有机化学中会发生什么
- 这些分子是共轭的吗?
- 有机化学中的共轭与共振
- 成键和反键轨道
- 烯丙基阳离子、烯丙基自由基和烯丙基阴离子的分子轨道
- 丁二烯的分子轨道
- 二烯反应:1,2和1,4加成
- 热力学和动力学产物
- 更多在1,2和1,4添加到双烯
- 顺式和反式
- Diels-Alder反应
- Diels-Alder反应中的环二烯和亲二烯
- Diels-Alder反应的立体化学
- Exo vs Endo产品在Diels Alder:如何区分他们
- Diels Alder反应中的HOMO和LUMO
- 为什么Endo vs Exo产品在Diels-Alder反应中更受青睐?
- Diels-Alder反应:动力学和热力学控制
- 复古Diels-Alder反应
- 分子内Diels Alder反应
- Diels-Alder反应中的区域化学
- Cope和Claisen重排
- Electrocyclic反应
- 电循环环开启和关闭(2)-六(或八)π电子
- Diels Alder练习题
- 分子轨道理论实践
19芳香分子的反应
- 亲电芳香族取代:简介
- 亲电芳香族取代反应中的激活和失活基团
- 亲电芳香族取代反应机理
- 亲电芳香族取代中的邻位、对位和元位董事
- 理解Ortho, Para和Meta director
- 为什么卤素是正对位的?
- 双取代苯:最强电子供体“胜出”
- 亲电芳香族取代(1)-苯的卤化
- 亲电芳香族取代(2)-硝化和磺化
- EAS反应(3)- Friedel-Crafts酰基化和Friedel-Crafts烷基化
- 分子内Friedel-Crafts反应
- 亲核芳烃取代(NAS)
- 亲核芳香族取代(2)-苄基机制
- “苄基”碳的溴化与氧化反应
- Wolff-Kishner, Clemmensen和其他羰基还原
- 芳香族侧链上的更多反应:硝基还原和拜耳Villiger反应
- 芳香族合成(1)-“操作顺序”
- 苯衍生物的合成(2)-极性反转
- 芳族合成(3)-磺酰基阻滞基团
- 桦树减少
- 合成(7):苯及相关芳香族化合物的反应图谱
- 芳香族反应与合成实践“,
- 亲电芳香取代的实践问题
谢谢! !
我也很困惑这个问题。
现在我明白了!:)
有没有办法在羧酸上加烷基呢?假设我想从羧酸变成叔取代醇。
如果你使用有机锂试剂,你可以把烷基加到羧酸上形成酮。它们也会使羧酸脱质子生成羧酸盐,但它们的能量足够大,可以加在一起形成碳碳键。然而,我相信形成的中间体是相当稳定的,在检查之前不会消除酮。要得到三醇,你必须把酮交给另一种等效的有机锂(如果你喜欢,也可以是格氏)。希望这能回答你的问题。
坦克,我很困惑
大多数研磨与碳水化合物反应。酸吗?
如图所示,它们通常是质子化的!
谢谢它对我的帮助
羧酸+胺是另一个常见的质子转移的例子,学生们错过了。我不知道我见过多少RC(=NR)OH产物——很多。
谢谢你的帮助那么如何从羧酸中制备酯而不进行费舍尔酯化反应呢?
啊,那种尴尬的感觉,当你意识到你忘记了几天前你还很熟悉的东西。
嗨,詹姆斯!
很棒的帖子,当我上学期看到它时,我已经理解了它,但最近我看到了你在评论中提到的有机锂反应。我有点困惑,为什么如果你加入两个等量的有机锂你可以用第一个去质子酸,第二个来加但你不能用两个等量的格氏试剂做同样的事情。仅仅是因为格栅变弱了吗?我一直认为这两种试剂都是非常强的亲核试剂/碱,没有太大区别。
——困惑但希望得到启迪(弗兰克W.)
我是马特,我是有机大师的导师。坦率地回答你的问题,我首先同意,乍一看,格氏酸会简单地去质子化羧酸,而烷基锂通过连续的去质子化/加成不可逆地形成一个锂离子,这似乎有点疯狂。在大多数情况下,格氏和烷基锂的反应方式相同,尽管我注意到,烷基锂在以脱质子化为具体目标时更常用(例如,形成叶立德)。如果你研究了足够多的机制,你最终会遇到许多这种令人沮丧的不一致的例子,必须通过一些挥手来合理化。可能没有人知道确切的答案,这可能与其中任何一个有关:
1)在此背景下,Li+阳离子的lewis酸略强
2) C-Li键的亲核性略高于C-Mg键
这可能是一种过度简化,我欢迎任何知道实验数据或计算研究的人来解释这一点。机制本身可能完全不同,因为这些试剂有时会做一些奇怪的事情,比如单电子转移(SET)自由基机制。
马特-我想说你的答案(1)是好的,但你的答案(2)是多余的。根据定义,如果R-Li反应性更强那是因为它有更高的亲核性
但是回到李路易斯酸度。你可以把它和LiAlH4和NaAlH4或者NaBH4和LiBH4的差异进行比较。在这两种情况下,锂盐的反应性更强。很多差异可能是由于刘易斯酸度,但也可能有溶剂化的差异,影响反应性。事实上,溶剂化/Lewis酸性在很大程度上解释了LAH和NaBH4反应性的差异。对裸离子AlH4-和BH4-的热化学研究表明,它们的氢化物结合能没有差异,在无溶剂和反离子的情况下,AlH4-和BH4-还原CO2的活化能相同。
思考有机金属或有机溶液中的无机盐等物质的结构并不总是那么容易。即使是乙醚和四氢呋喃结构上的细微差异也会改变溶剂化性质,这些东西会对反应性产生很大影响。幸运的是,我们有像Hans Reich和Dave Collum这样的人来帮助我们更好地理解它。
谢谢你!我的疑问解决了:)
伟大的解释!
grignards试剂会和RCOCl中C=O旁边的酸性氢反应吗?
一般不会,除非酸氯化物受到特别的阻碍。C-H键不像O-H键那么快地脱质子,因为1)键必须重组(再杂化),2)C-H键的酸度取决于它与羰基的排列。
还有哪些官能团会引起类似的问题?
羟基(OH),硫醇(SH),以及其他一般的弱酸性官能团。羧酸是迄今为止最常见的情况。
请说明:格氏试剂+羧酸氯- >酮+ mg -卤化物。
C-Cl键的反应性是否如此之强,以至于不能进行简单的格氏质子化反应?
"羧酸氯"的意思是,"酸氯"实际上不含酸性氢。所以格氏质子化在这里不是问题,虽然羧酸是问题。
你能解释一下为什么HBr在亲核加成过程中不加到羰基上吗?
碳溴键比碳氧键弱得多。即使形成了C-Br键,它也会很快被排出,重新生成羰基。
谢谢
亲核试剂不能加入HCOO-为什么?即使它不与格氏试剂反应
羧酸盐的电荷实际上在两个氧之间,这可能解释了为什么它不喜欢表现得像羧基化合物。
是的,没错。带负电的氧是强的给体而羰基碳的亲电性较弱。
我在解题时太困惑了。这篇文章说得很清楚。
谢谢。
我的困惑现在完全消除了
...
用格氏试剂制取酒精