烯醇、烯醇化物
醇醛加法和缩合反应
最后更新:2022年12月6日|
醇醛加成、缩合反应(Base-Catalyzed)
的醇醛加成反应是添加一个烯醇化物到一个醛(或酮)。加热与基地可能导致损失的水给一个新的碳碳π键,给产品我们称之为醇醛缩合产品。
类似的反应可以继续其他物种的烯醇化物。他们通过不同的名称,但基本过程是相同的。
表的内容
- 醇醛反应(也称为“醇醛加成反应”)
- 醇醛加成反应的机理
- 醇醛缩合
- “穿越”醇醛反应,
- 醇醛酮的反应
- 分子内醇醛反应
- 醇醛反应反过来——“复古醇醛”
- 总结
- 笔记
- 好处:更多关于消除步骤
- 好处:表亲的醇醛反应(Knovenagel,亨利,Reformatsky)
- 测试你自己!
- (高级)引用和进一步阅读
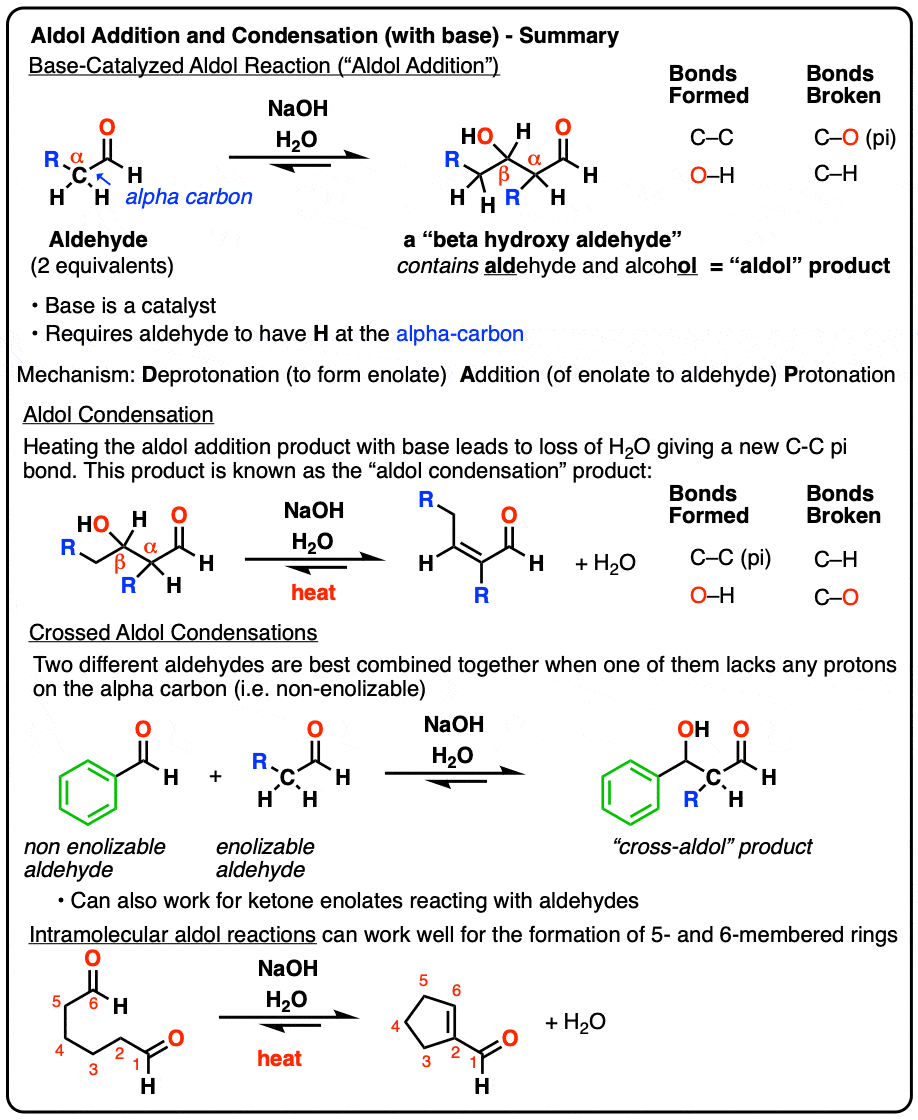
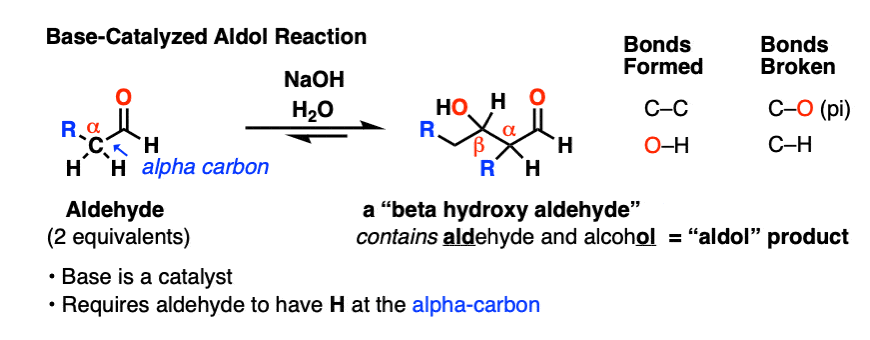
1。醇醛加成反应
当某些醛强碱处理,两个摩尔当量的开始醛可能形成新的碳碳键。由此产生的产品,一个“beta-hydroxy醛“既包含一个“肾上腺脑白质退化症”ehyde和alcohol。
除了新的碳碳键,一个碳氢键坏了,就像一个切断(π)债券:
这种反应被称为醇醛反应或“醇醛除了反应”,以区别于水的情况下失去了给一个新的碳碳双键(醇醛缩合,明白了第3部分)
这种类型的反应也可以用于酮(见下文)和其他官能团(见附录)。
在本文中,我们将关注base-catalyzed版本的醇醛。一个催化版本也存在;这是讨论(参见:催化醇醛反应]
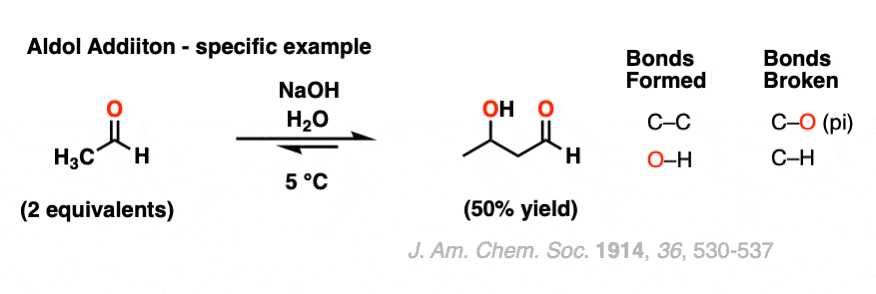
这是一个具体的例子,一个醇醛加成反应,使用2的乙醛(又名“乙醛”)。
2。醇醛加成反应机制
所以这个反应是如何工作的呢?基地的目的是什么?和新碳碳键是怎样形成的呢?
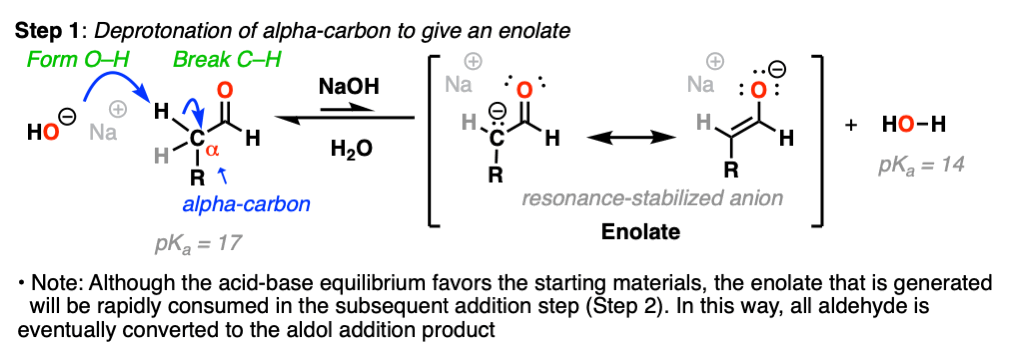
思考机制的样子时,首先要注意的是,我们使用的是强碱(氢氧化钠)。
这就引出一个问题:什么是最酸性质子吗?提示:这不是碳氢键上醛!(这将导致一个酰基阴离子,这不是共轭)上。
看着这些债券形式和决裂,你应该能够看到我们打破一个碳氢键碳直接相邻醛羰基(即“α碳”)。
删除此碳氢键由强碱共轭系统的结果烯醇化物离子。(步骤1,形成地,打破碳氢键)。
因此,醇醛反应是必然要求醛是enolizable(即有一个质子α碳)。没有质子→烯醇化物→无醇醛。
悬停看到enolizable vs非enolizable醛的一些示例或点击这个链接。
{kind=link}
注意酸碱平衡实际上倾向于起始原料——我们使用氢氧化钠(共轭碱的水,pK一个= 14)做出了更强大的基础烯醇化物,共轭碱乙醛,pK一个= 17)。
的反应的关键烯醇化物,一旦形成,是一个很好的亲核试剂并能迅速与任何可用的反应亲电试剂挂在解决方案——比如,例如,醛。
需要我们的产品形式碳碳和打破切断(π)。
的烯醇化物因此增加了醛在步骤2(形成碳碳,打破切断(π)),最经典的机械的步骤的一个例子金属羰基合物,加法。
通过这种方式,所有的开始醛最终转化为醇醛添加产品,尽管不利的酸碱平衡。
后加成反应,生成的负电荷在氧气然后经历质子化作用的溶剂(步骤3,形成地)。
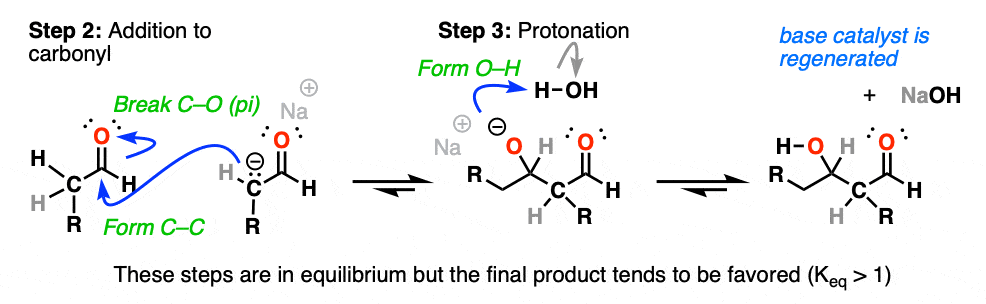
如果你以前覆盖醛和酮的反应,希望这一系列事件看起来很熟悉。
这是另一个经典的例子两步机制(addition-protonation)醛,烯醇化物作为亲核试剂。(如果算好,好- 3步骤烯醇化物形成)。(看:醛和酮- 14反应遵循相同的机制]
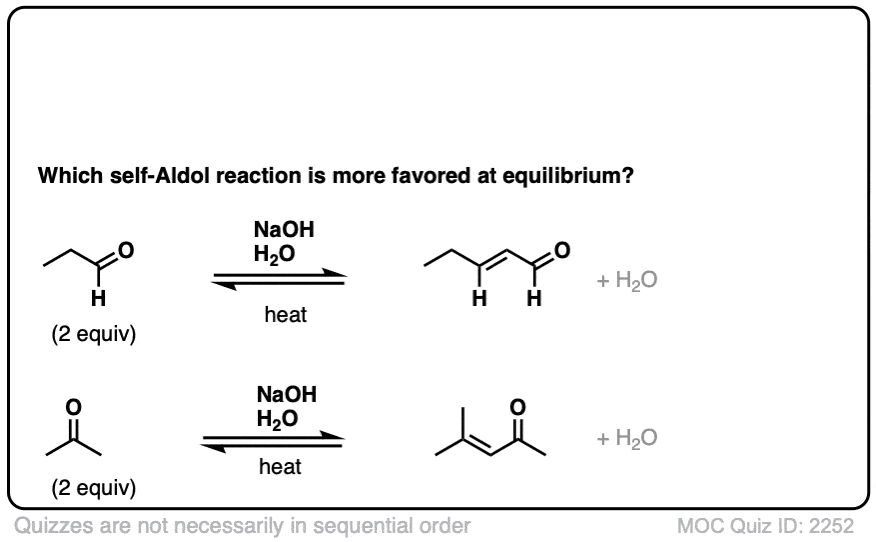
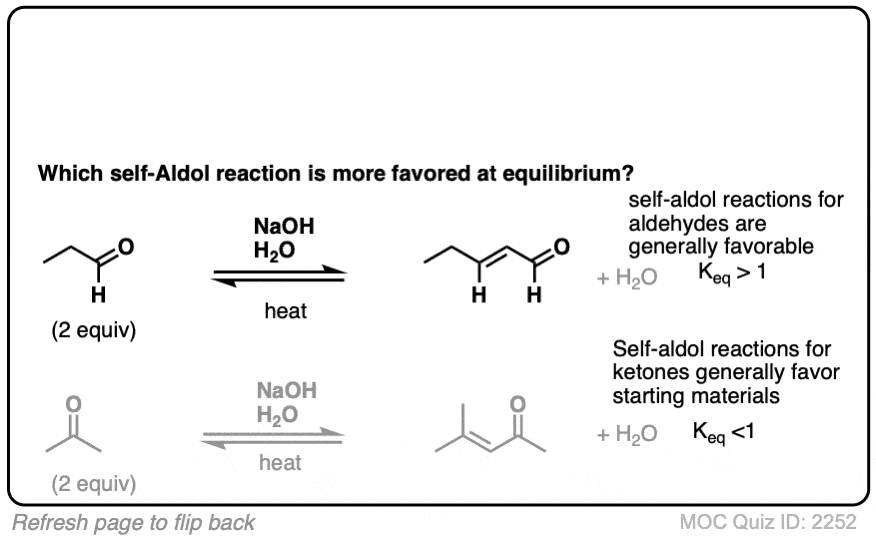
在这些条件下,注意所有3步骤处于平衡状态。与醛、平衡通常更喜欢最后的醇醛产品。(注1]
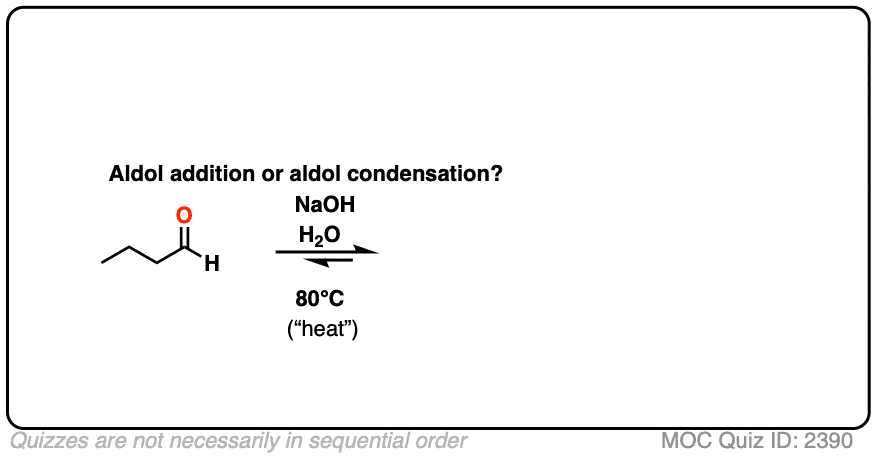
3所示。醇醛缩合反应
但这还不是全部!有更多的醇醛比只是一个无聊的老了加成反应。
如果由此产生的醇醛(beta-hydroxy添加产品醛与基地)加热,它会失去水给一个新的碳碳π键。
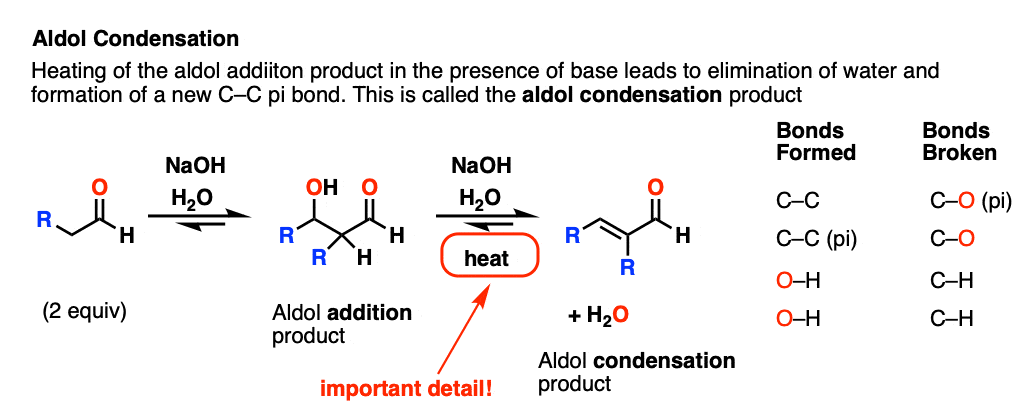
我们破碎的碳氢键,断了噢,形成碳碳(π)。
这被称为一个醇醛缩合反应因为水是形成一个副产品。
对我们来说最重要的区别因素是“热”,它倾向于消除反应。(看到的:消除反应的热量]
写给每个人读这他预计测试这种材料在考试中:如果你看到“热”,假设您将画出缩合产物。
两个经典的“真实”的例子,比较主要产品为同一反应在低温(0 - 5°C)和“高温”(80°C)。
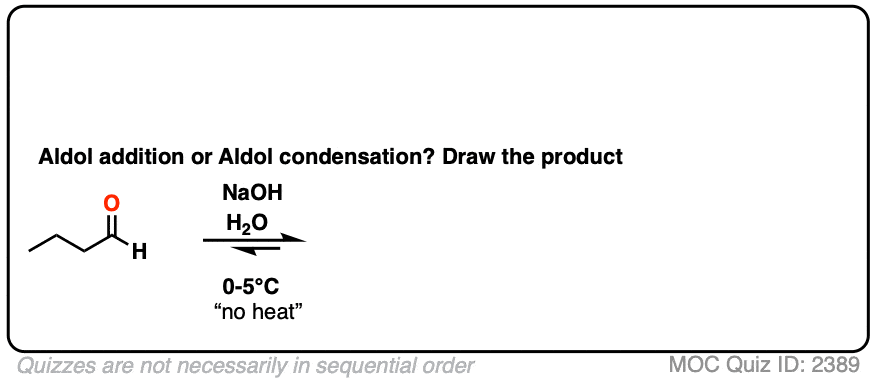 点击翻转
点击翻转
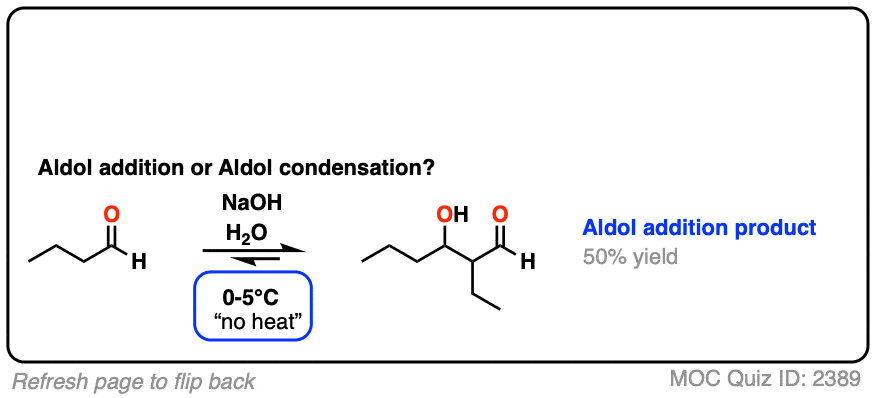
 点击翻转
点击翻转
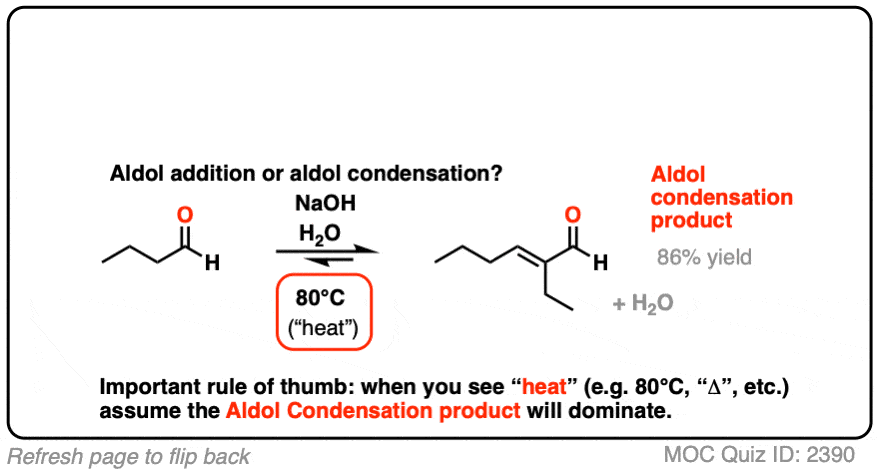
(见注2供参考)
在现实世界中,还有其他的因素可以决定产品分布,但“热”是最重要的一个“有机化学概论”的观点。(注3]
消除步骤是这样运作的一个例子E1CB反应——不是一个E2!更多的讨论见附录(注意4。]
4所示。“穿越”醇醛反应
到目前为止,我们只处理“self-aldol”反应,两个相同的等价物醛结合新产品。
让我们来看下一个合乎逻辑的步骤。为什么不结合两个呢不同的醛吗?
例如,假设我想要以下两个醛产品正确的通过醇醛的反应。
所以我添加两个醛溶液,添加我的催化基础,把我的脚,等待反应完成,相信宇宙中,它将所有的工作。
什么可能出错。正确的吗?
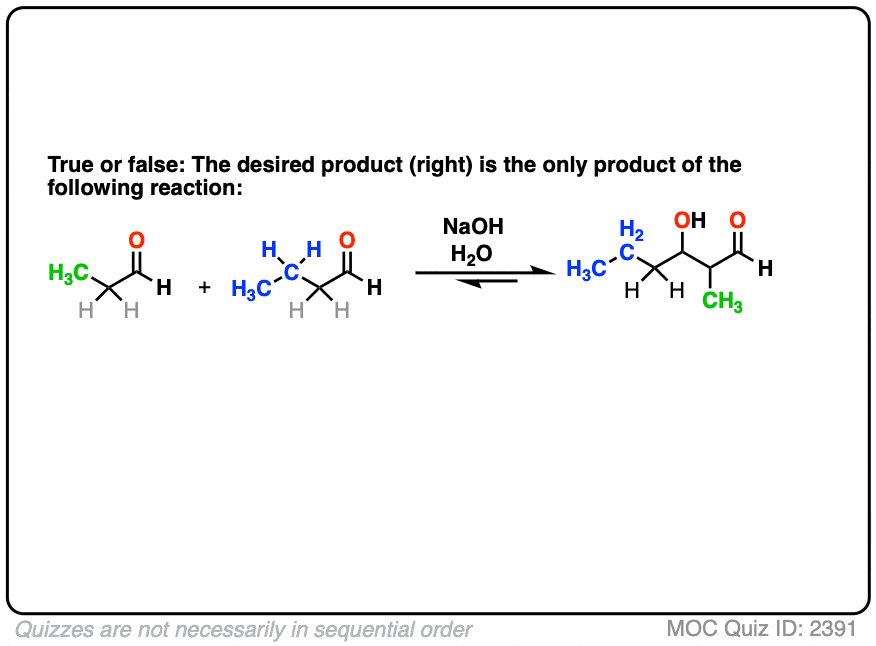 点击翻转
点击翻转
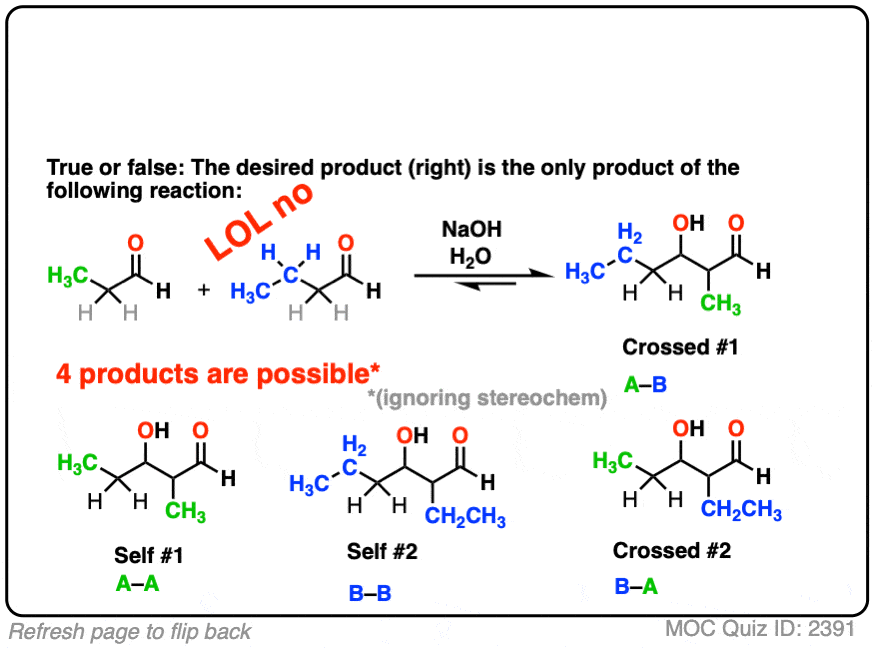
当然只是在开玩笑。它是化学。很多事情会出错!
首先,如果我们有两个enolizable醛在解决方案,那么我们可能会形成两种不同的烯醇化物。
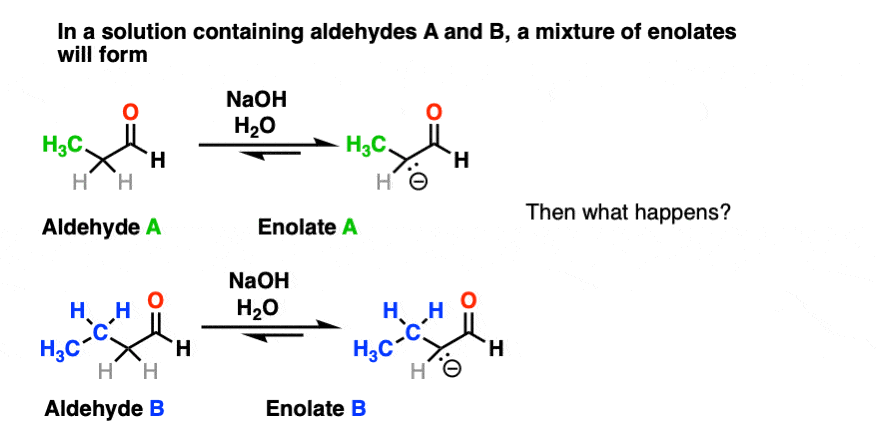
其次,由于我们使用催化基地,有大量的醛。
每一个烯醇化物反过来,可能与其中一个醛反应,给两个“self-aldol”产品和两个“交叉醇醛”产品。
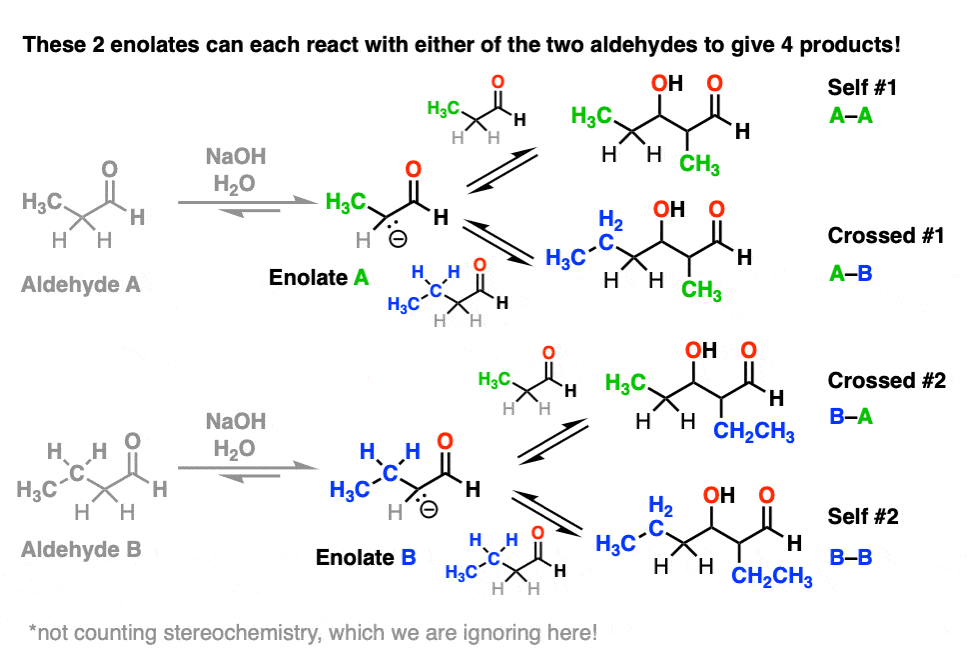
并不是所有的人会形成数量相等,请注意,但这仍然是4的产品。
(我们使事情更容易通过选择忽略立体化学。(注5]
这个结果是化学家在业务可能会调用BFM的反应。委婉地说,这代表该死的大混乱。虽然这个真实的例子(从1914年)实际上变成了好:(注6]
悬停看到一个现实生活的例子或点击这个链接。
{kind=link}
事实是,没有人想要运行一个反应生产4种不同产品的重大收益。混合物的分离是一个巨大的时间杀手,每一个化学家试图避免尽可能多。
怎么能这样做更有效?
一种方法是使用非enolizable醛合作伙伴的反应。只有一个烯醇化物亲核试剂担心的。
这使得醇醛反应更可预测的,特别是如果我们使用过多的非enolizable醛self-reaction降到最低。
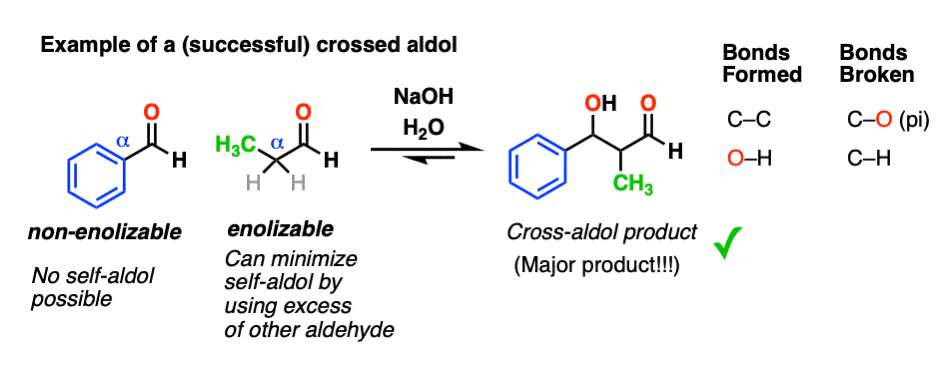
5。醇醛酮的反应
烯醇化物也可以形成的酮在醇醛,酮也可以亲电试剂反应。
这是self-aldol反应丙酮为例。
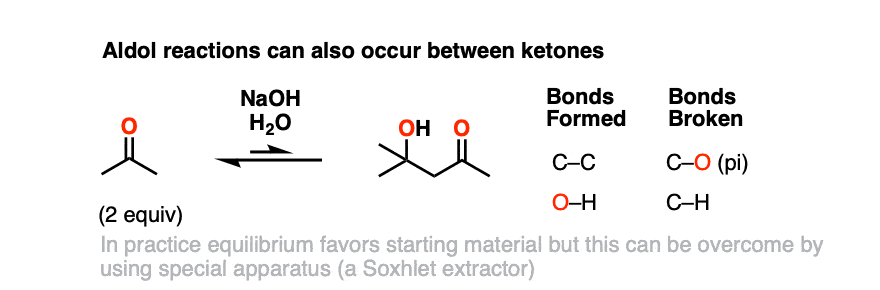
在实践中,self-aldol丙酮工作并不好——平衡倾向于起始原料,但使用一个设备被称为索氏提取器,可以隔离产品的合理收益。
当我们的话题值得注意的是,酮是generally活性低于醛的亲核试剂,因为羰基碳是更多的位阻,也更富电子(烷基团体electron-releasing——回想一下,他们是“催化剂”傅克反应)。
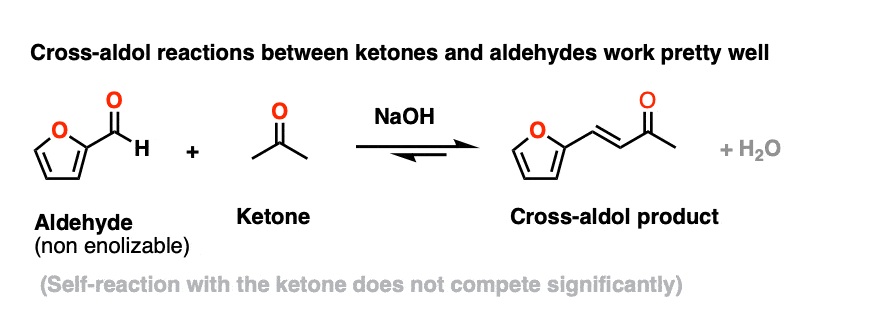
然而,使用酮烯醇化物(由基地)亲核试剂与非醇醛反应enolizable醛非常好用。诸如此类的例子还有很多,这是由其自己的名字-施密特克莱森缩合,不需要知道。
这里有一个例子从有机合成。(Self-aldol丙酮不竞争的任何重要程度)
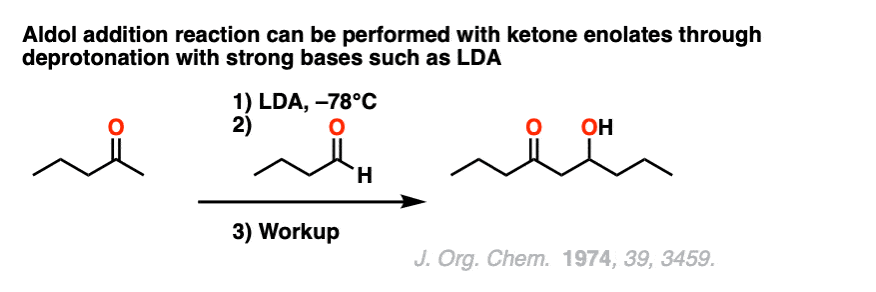
做醇醛反应的另一种方式酮烯醇化物是pre-form烯醇化物与一个强大、bulkyl基础等锂diisopropylamide(乔治。),然后添加醛。
(请注意7]
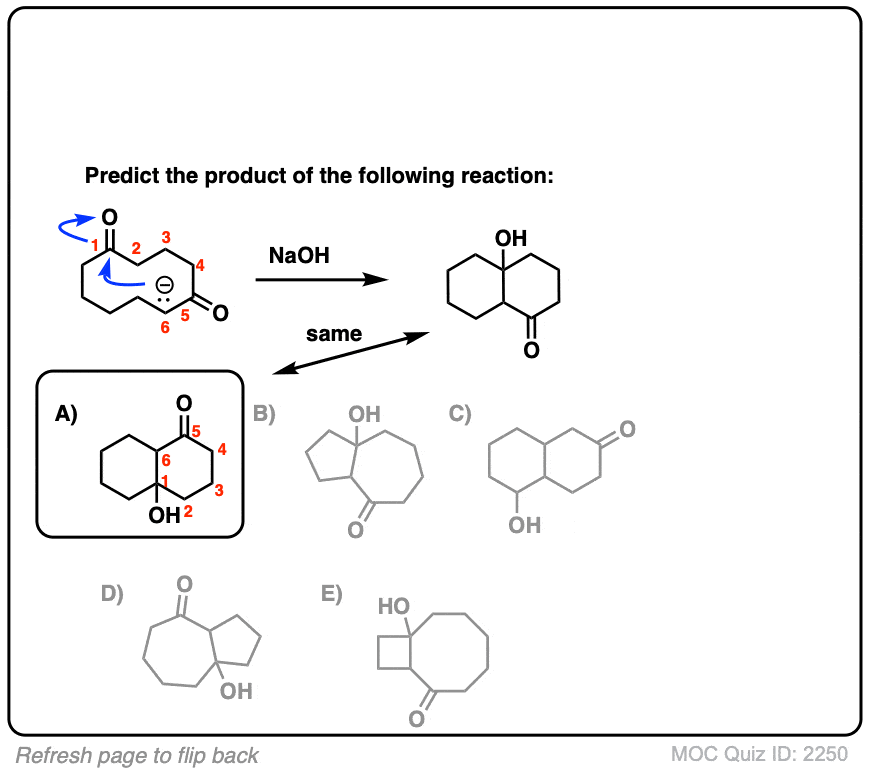
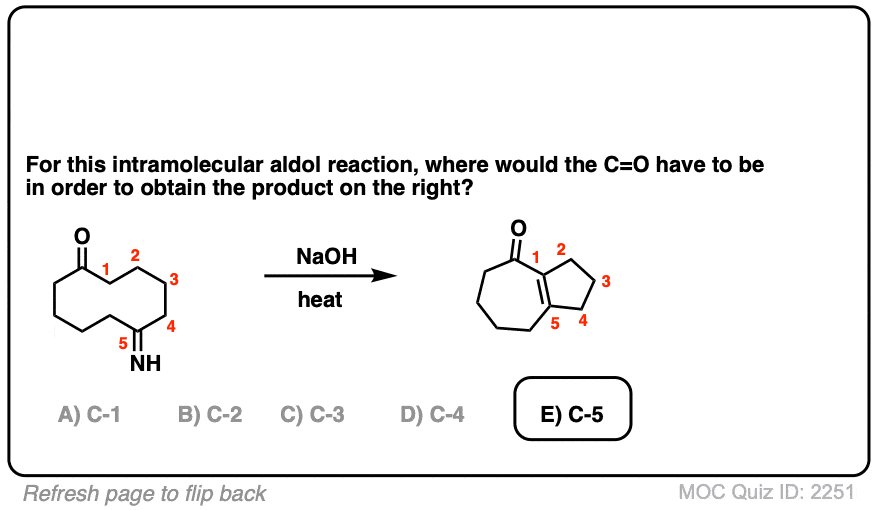
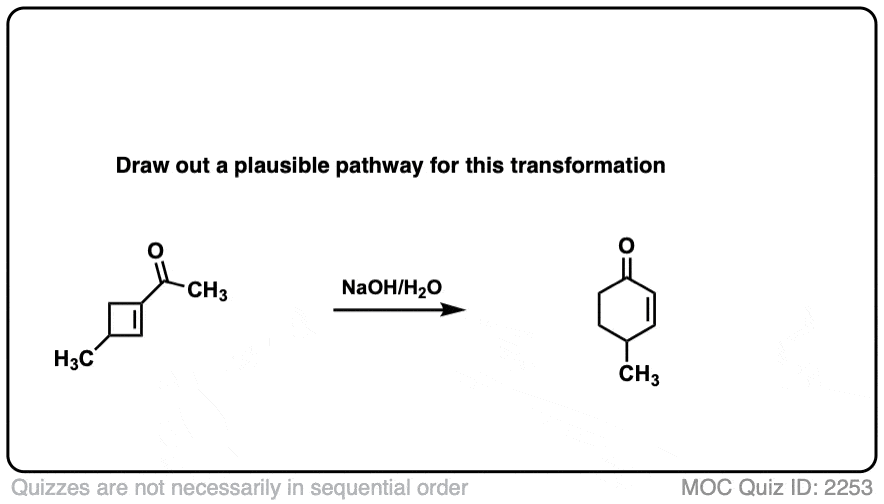
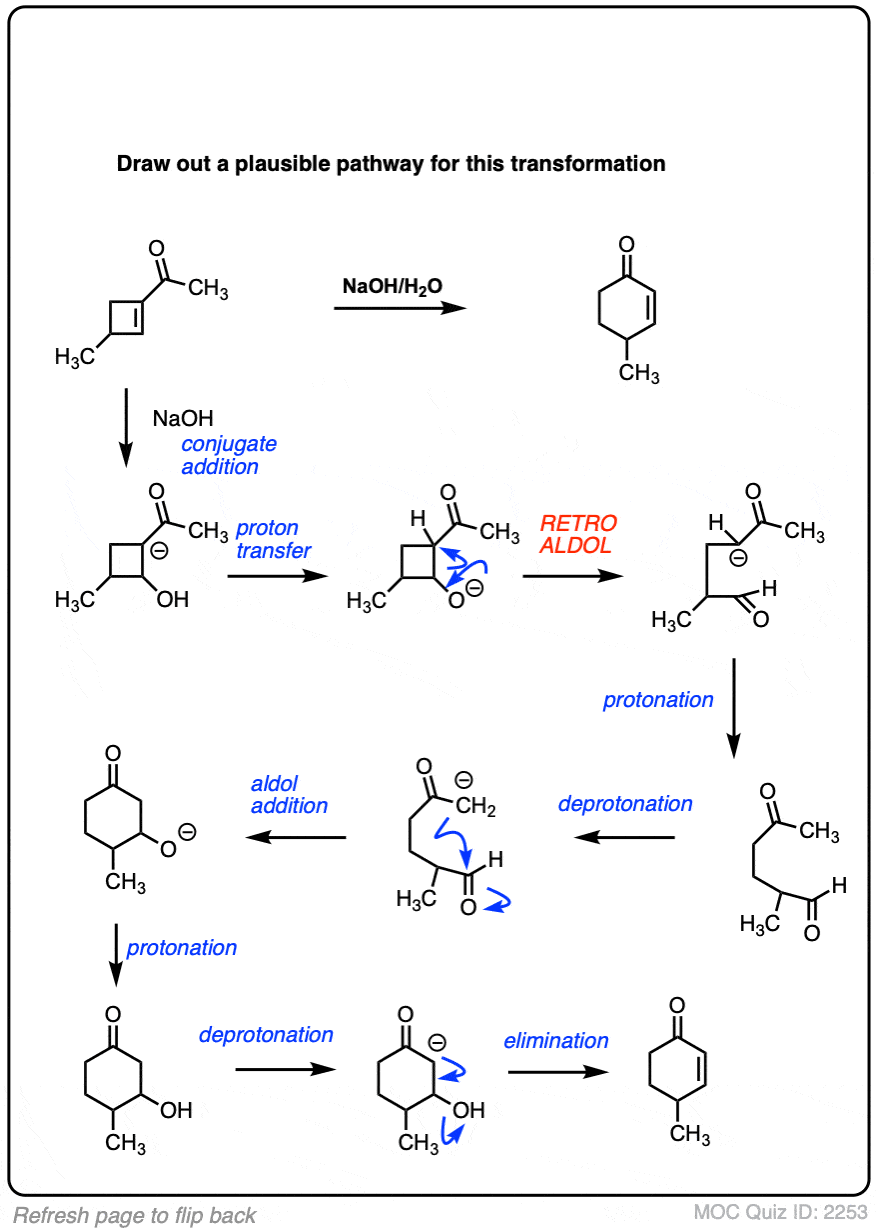
6。分子内醇醛反应
我总是告诉学生期待看到分子内的版本的反应在考试,因为老师知道他们是一个伟大的方式测试你理解一个给定的反应没有引入任何新概念。
所以我将过失如果我省略的例子分子内的醇醛反应,工作最好当5 -或6 -元环可以形成。(提醒——5和6元环最环的压力)。
看看这个例如:
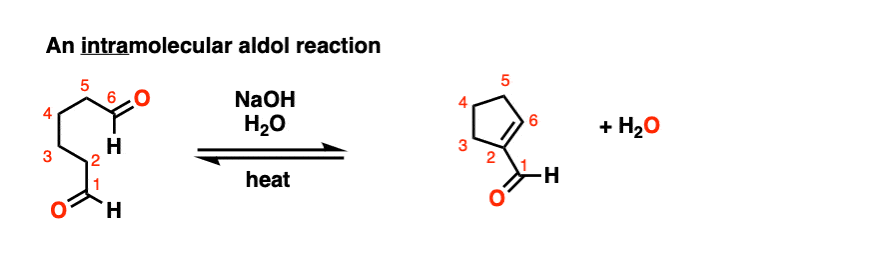
(裁判)注意没有区别在债券形式或打破分子间的情况下(形成碳碳,打破切断(π),打破碳氢键)。新成立以来烯醇化物和醛在同一分子,我们必然会形成一个环。
{kind=link}
分子内醇醛密集的一个常见的应用是在罗宾逊环状结构的第二步反应。
[也:看到帖子:罗宾逊的环状结构]
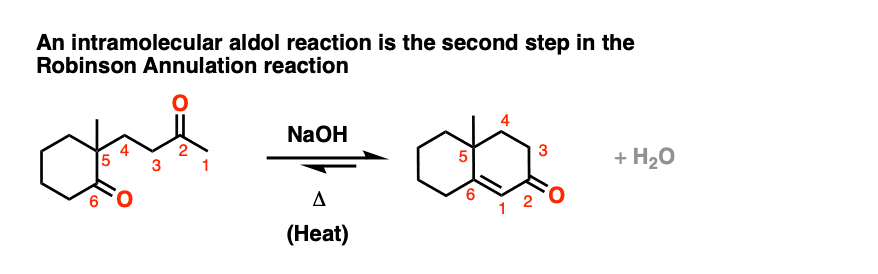
几个例子的分子内醇醛反应在“测试自己”一节。
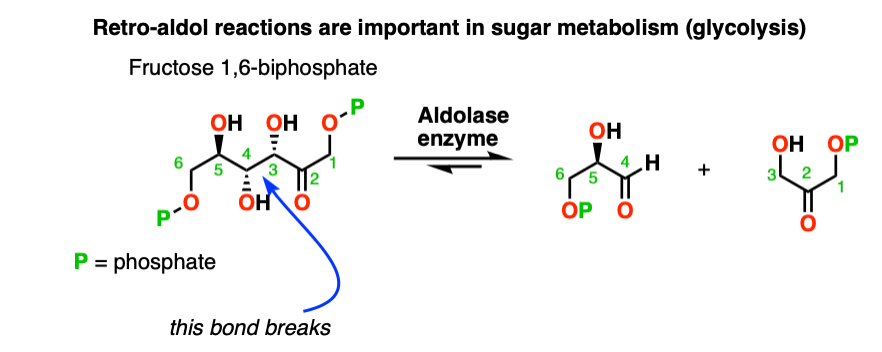
7所示。Retro-Aldol反应(醇醛反应的可逆性)
醇醛的反应是一个平衡。通常与醛的反应倾向于最终产品与原料。
但是有时可能使反应逆向工作。
这是有时被称为retro-Aldol的反应。这里的一个例子retro-aldol反应由释放环应变驱动的
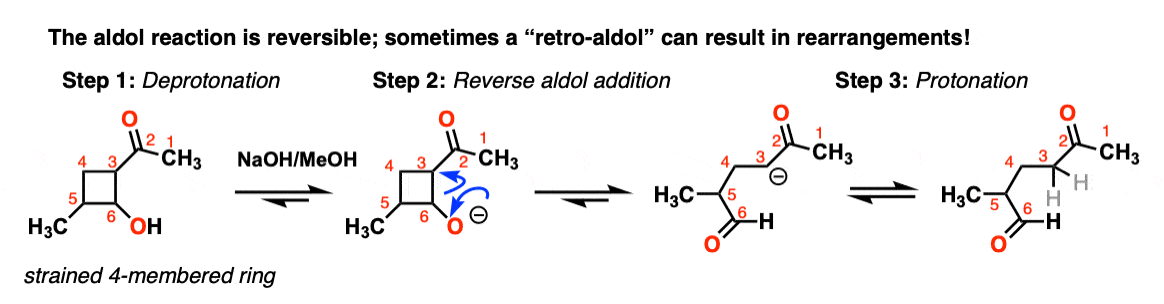
(注意,最初的原始材料是没有通过一个醇醛)。
甚至有酶(称为“醛缩酶”),可以促进retro-aldol反应,比如在糖酵解。(注8]
8。总结
- 的base-catalyzed醇醛除了反应是添加一个烯醇化物到一个醛(通常)。
- 如果采用热,期望进一步损失的水和形成新的双键,导致醇醛冷凝产品。
- Cross-aldol反应效果最好,当一个人enolizable醛随着使用非吗enolizable醛随着亲电试剂。酮烯醇化物也极好的醇醛的亲核试剂反应。
- 酮的self-aldol反应可能发生,但他们不太有利比醛(产品更多的位阻)。
- 当心分子内醇醛缩合反应,特别是5 -或6 -元环可以形成。
- “Retro-aldol”beta-hydroxy酮的反应是破碎或恢复beta-hydroxy醛烯醇化物/醛起始原料。
- 醇醛反应也可以被酸催化,通过一个收益烯醇中间。关于这一点,在这里看到的。
笔记
注1。一项研究测量了平衡常数K 400 (裁判]。平衡常数的位阻降低产品(如增加醛变得更加扩展)。
注2。的第一反应是江淮1914年36岁,530年。第二反应是格氏& Vestermann牛化学。Soc。Fr。第4部分,1925年37岁的425年。在网上很难找到,但在引用全面的有机合成。
注3。当一个芳香醛使用很难使反应停留在添加阶段,随着双键将通过与芳环共轭相当稳定。
注意4。在最近的一项研究机制的醇醛缩合,损失的离去基团(-)决心速率决定步骤。(裁判]。
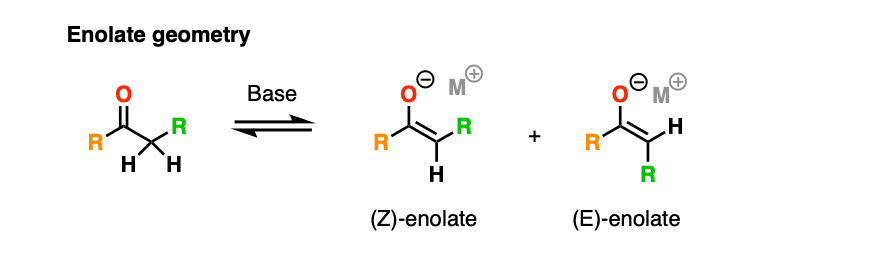
注5。我们省略立体化学在这篇文章中,让事情变得简单。我也选择画出烯醇化物作为一个负碳离子的(正确的)平面几何与一个负电荷的氧气。
与烯醇化物这种方式,一个特别重要的细节的几何烯醇化物,E或Z。
1957年,齐默尔曼和Traxler提出了一个模型来预测醇醛的立体化学(及相关反应)通过一个封闭,六元比作过渡态金属米坐标的氧气烯醇化物和醛。
在这个模型中,最低过渡态是一个笨重的R组占领赤道在“椅子”。
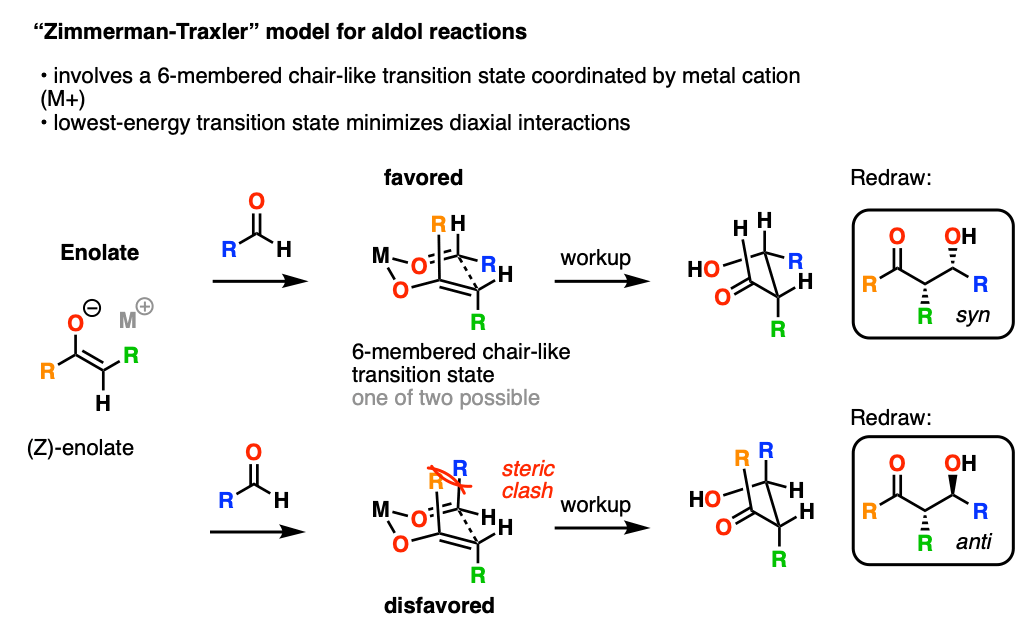
对立体选择性金属M有巨大的影响。多米尼加债券越短,越压缩“椅子”过渡态,和更大的立体选择性。特别是,醇醛反应与硼烯醇化物显示壮观的立体选择性。
注6。这个例子从1914年开始,在现代光谱和分离技术识别所有可行的反应产物。的主要产品是蒸馏54%的收益率,这实际上是一个很好的结果!老概括称为“爱规则”说的主要醇醛产品结果附件“替换”烯醇化物“替换”醛。
请注意7。这是一个典型的例子一个醇醛之间的反应酮烯醇化物和一个醛使用锂di-isopropylamide (乔治。),形成“替换”烯醇化物。(裁判]
注8。复古的醇醛反应实际上是糖代谢的一部分!在体内,1磷酸二氢6-fructose通过行动的醛缩酶裂解,使磷酸甘油醛3 -磷酸和dihyroxyacetone。
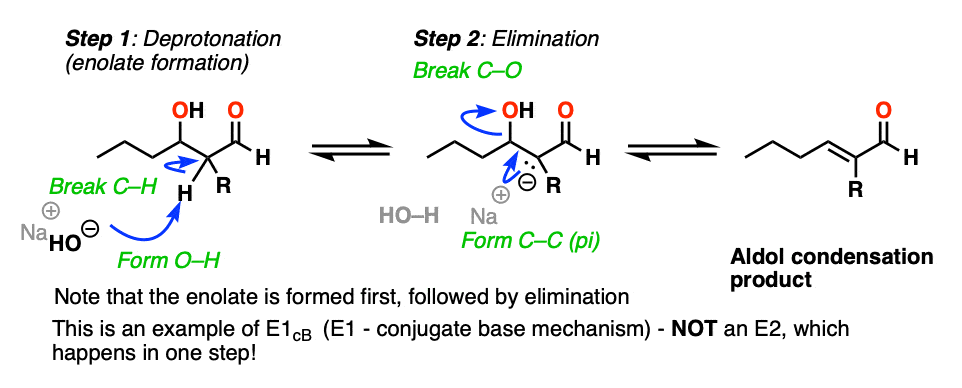
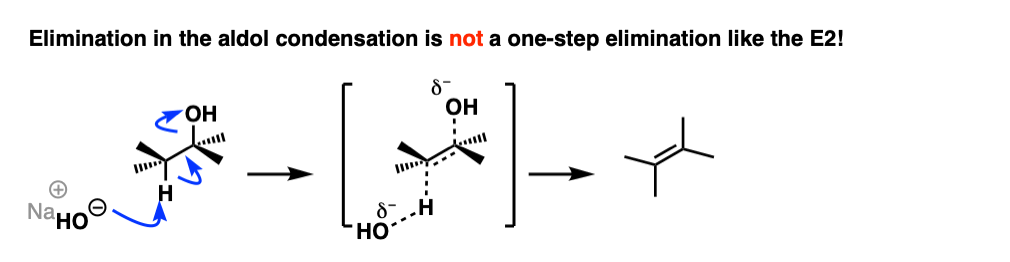
消除一步醇醛缩合反应
所以“消除”步骤是如何工作的呢?
首先,为什么HO (-)离去基团吗?难道不是一个强大的基础,因此坏离去基团吗?是的,它是一种强碱,通常不是一个好离去基团在消除反应。然而,这不是一个典型的消去反应。
第一个本能可能会认为这是一个E2反应,因为我们使用强碱。
这不是一个E2反应,因为它是不协调!
这是一个例子E1CB(消除,单分子的速率决定步骤,通过共轭碱)。(看到帖子:E1cB机制]
在强碱(如氢氧化,共轭碱的水,pK一个14),烯醇化物可以由beta-hydroxy吗醛(pK一个约17 - 18)。
请注意,烯醇化物产品比氢氧离子实际上是一个强大的基础,所以平衡起始原料的躺在一边。
的烯醇化物是一个中间,可以观察到的和孤立的(不同于双分子的过渡态的E2!)
现在,如果你认为负电荷被困在p轨道上的碳,想象p轨道重叠与相邻切断σ*键。如果重叠是充分的,那么债券可以形成新的碳碳(pi)与c键的断裂。
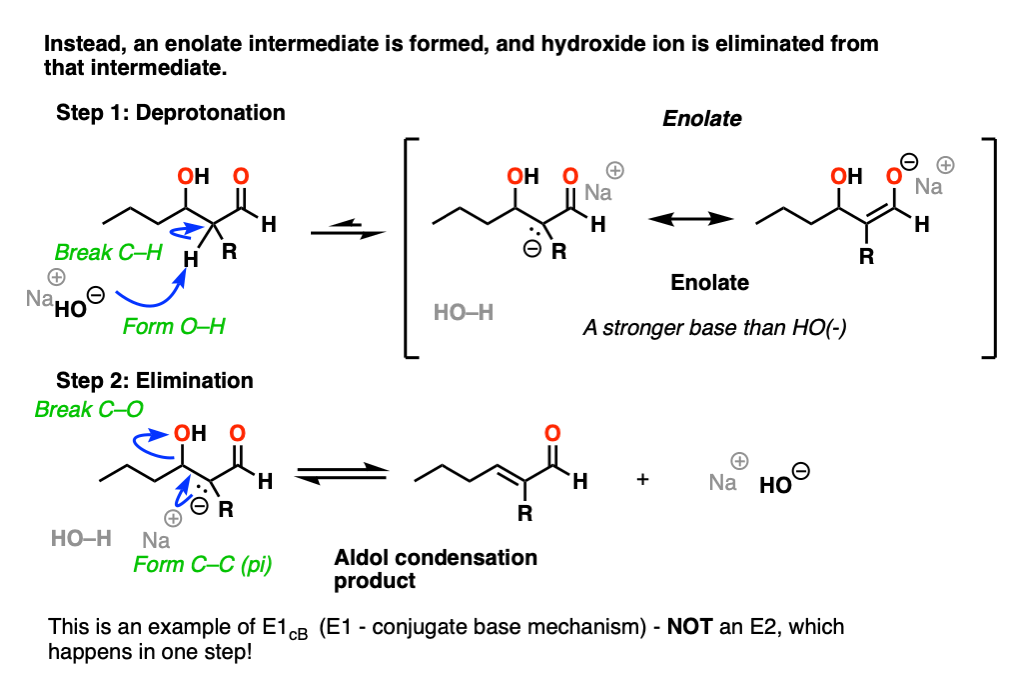
所以在第二个步骤中,我们会从一个强大的基础(烯醇化物)较弱的基础(氧化)。
是的,氢氧根离子是一种强碱和坏离去基团,但实际上驱逐氢氧化钠的反应是支持从酸碱的角度(强酸+强碱- >弱酸性+基地)。
此外,产品有了一个新的共轭双键,C = O键,这是稳定。
醇醛反应的表亲——Knovenagel,亨利,Reformatzky和其他变体
在本文中,我们已经讨论了醛和酮的烯醇化物的反应,但也有可能对其他“烯醇化物”形式。这应该是一个单独的文章,但几乎都是非常相似的机制的醇醛所以他们对待短暂。(步骤1 -形成烯醇化物;步骤2 -之外羰基,然后第三步——质子化作用,其次是消除在某些情况下)。
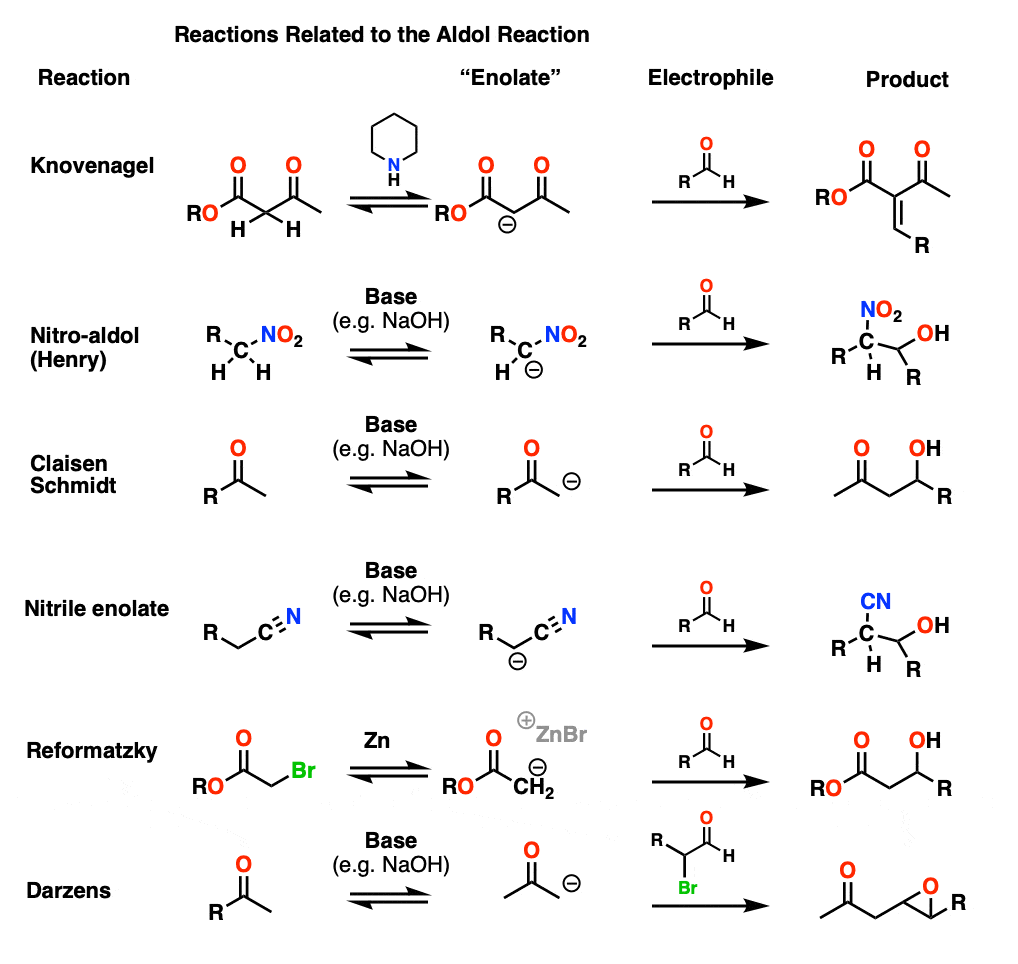
Knovenagel凝结
beta-keto Knovenagel的凝结酯(或类似的)复合处理轻微基地(经常使用哌啶)形成烯醇化物然后,它可以攻击醛。损失的水通常是快速通常由于α质子酸(pH值约12)。
亨利的反应
亨利nitro-aldol或反应,碳毗邻NO2组是deprotonated让一个物种一样烯醇化物。
Claisen-Schmidt
Claisen-Schmidt(名字不重要)酮烯醇化物形成和增加一个吗醛。
腈烯醇化物
相邻的碳腈也可能是deprotonated虽然pK吗一个大约30,使用氢氧化钠是一段。本质上这是没有不同于典型的醇醛的反应。
Reformatsky反应
alpha-bromo Reformatsky,一个开始酯然后把它与细锌粉。这是很像形成格氏试剂,除了产生的负碳离子是一个烯醇化物!的烯醇化物可以添加到一个醛或酮。
Darzens反应
这是一个稍微有点奇怪,一个醇醛alpha-halo醛作为亲电试剂。后添加烯醇化物,由此产生的醇盐可以形成一个环氧通过年代N2位移C-Br债券(见这篇文章环氧化合物的合成)。基本上醇醛反应haloether,然后拍照给一个关闭环氧。
测试你自己!
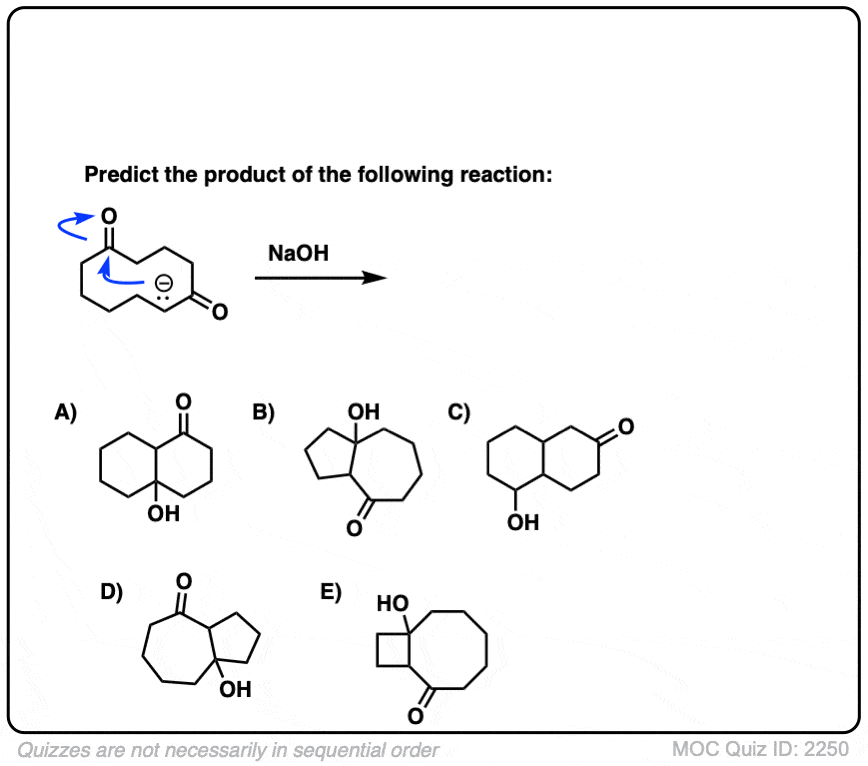 点击翻转
点击翻转
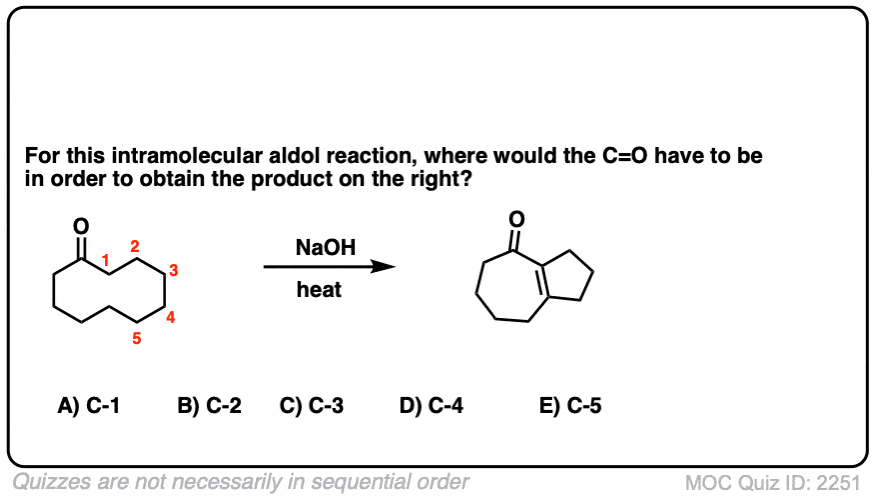 点击翻转
点击翻转
 点击翻转
点击翻转
 点击翻转
点击翻转
(高级)引用和进一步阅读
(高级)引用和进一步阅读
和审查全面的有机合成,卷2。页139 - 177是非常有用的在寻找有用的文献引用醇醛的反应。推荐。
- Valeraldehyd Ueber死Einwirkung des钠再见
a·鲍罗丁
j . Prakt。詹。1864年93年(1),413 - 425
DOI:10.1002 / prac.18640930168
第一个出版领域的醇醛的化学,由亚历山大鲍罗廷在1864年。本文涉及治疗戊醛(C5H10O)给我们现在称之为钠醇醛缩合产物。随后在1872年的一项报告涉及的治疗正丁醛与盐酸通过维尔茨获得产品类似报道。
这篇文章有历史细节鲍罗丁的丰富多彩的生活;除了化学,他也是一个受欢迎的作曲家,他的作品仍然是今天。 - 关于一个aldehyde-alcool
维尔茨,m·A。
牛詹。Soc。Fr。1872年,436年
没有DOI,但这是完整的链接
阿道夫·维尔茨研究了乙醛的治疗与盐酸在低温和获得一个新的“醛- - - - - -酒精“产品C4H8O2。“Ce陆战队est l醛-alcool,公平l 'objet de这个注意等我nomerai简称不相上下醇醛”。这个名字“醇醛”一直和我们在一起。 - Condensationen冯Ketonen麻省理工学院Aldehyden(冷凝与醛酮)
l·克莱森;Claparede,。
的误码率。1881年,14(1):2460 - 2468
DOI:10.1002 / cber.188101402192 - “Ueber死Einwirkung冯丙酮auf糠醛改Bittermandelol Gegenwart冯Alkalilauge”的影响丙酮糠醛和苦杏仁油(苯甲醛)的碱金属氢氧化物)
施密特,j·G。
化学。的误码率。1881年,14(1):1459 - 1461
DOI:10.1002 / cber.188101401306
之间的反应醛或酮有一个α-hydrogen芳香羰基化合物缺乏一个α-hydrogen叫做Claisen-Schmidt凝结。这个反应是命名的两个开创性的调查员Rainer路德维希·克莱森和j·g·施密特,他独立发表在1880年和1881年这一主题。 - 完整的醇醛缩合的机理
查尔斯·l·佩兰和Kuei-Lin张
《有机化学》杂志上2016年,81年(13),5631 - 5635
DOI:10.1021 / acs.joc.6b00959
一个很好的机械研究base-catalyzed交叉醇醛反应苯乙酮和苯甲醛之间,形成查耳酮。本文建立的最终消除HO(-)是通过观察病原反应步骤在这个特定的反应,反应速度更快2比H O2O(溶剂同位素效应)。 - 研究化合物auxin-a和auxin-b相关。第三部分。的制备和性质auxin-b cyclopentenyl模拟内酯
j·b·布朗、h·b·Henbest和e·r·h·琼斯。
j .化学。Soc。1950年,3634 - 3641
DOI:10.1039 / JR9500003634
一个分子内醇醛缩合的好例子。 - ”混合冷凝两酮很少制备有用。“从和c . h全面的有机合成卷。2。1.5节“醇醛反应:酸和碱催化”。页133 - 179。参见下面的格思里的论文,醇醛缩合平衡常数为0.039(即有利于原料)。可以得到不错的收益通过回收醇醛产品从基地索氏仪器。
- 乙醛的醇醛缩合反应的平衡常数和氢氧根Retro-Aldol催化反应的速率常数
j·彼得·格思里
可以。52岁的j .化学。2037年(1974)。f
DOI:https://doi.org/10.1139/v74 - 294
“间接热化学平衡常数的估算乙醛建议的醇醛缩合反应是少得多比一直相信…不可逆平衡常数为4.0×102米1…。焓的变化是-9.84千卡每摩尔。”(A calculation for ΔG° based on these parameters gives -3.55 kcal/mol]
醇醛缩合的类似的研究丙酮导致了平衡常数为0.039(即倾向于起始原料)和一个值为ΔG°+ 1.92千卡每摩尔。裁判。 - 伊万诺夫和Reformatsky反应的立体化学。我
霍华德·e·齐默尔曼和马约莉·d·Traxler
美国化学学会杂志》上1957年79年(8),1920 - 1923
DOI:10.1021 / ja01565a041
醇醛反应的Zimmerman-Traxler过渡态模型,这被证明是非常有用的在合理化(预测)立体化学的结果。 - 第一个直接和拆分Cross-Aldol醛的反应
Alan b .贝蒂和大卫·w·c·麦克米伦
美国化学学会杂志》上2002年124 (24),6798 - 6799
DOI:10.1021 / ja0262378
cross-aldol反应两种醛,穿过一个烯胺而不是烯醇化物中间,用氨基酸L-proline作为催化剂。
现实生活中的例子:
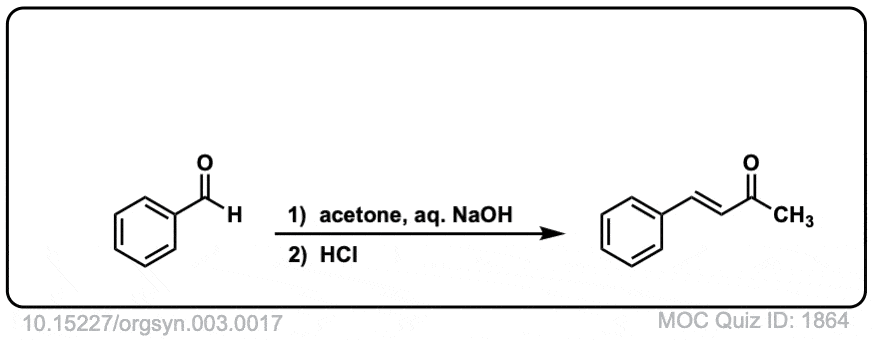
Org。Synth。1923年,17岁的3
链接DOI: 10.15227 / orgsyn.003.0017
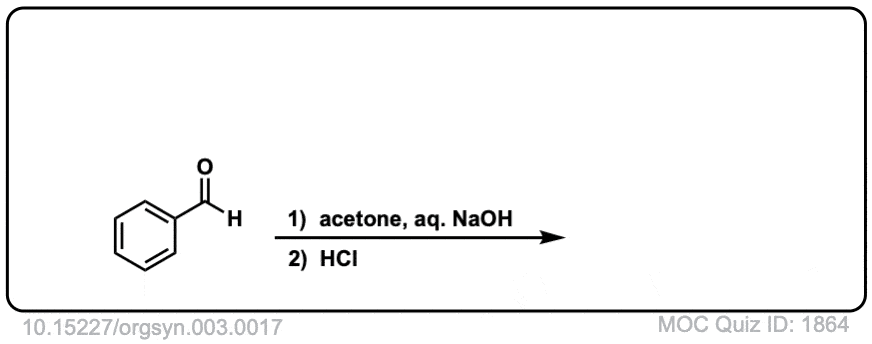 点击翻转
点击翻转
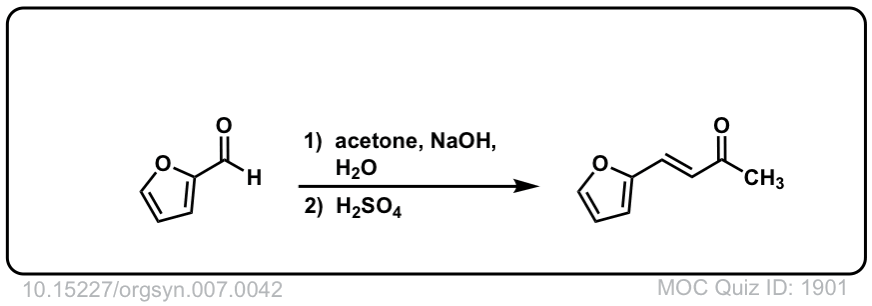
Org。Synth。1927年,7日,42
链接DOI: 10.15227 / orgsyn.007.0042
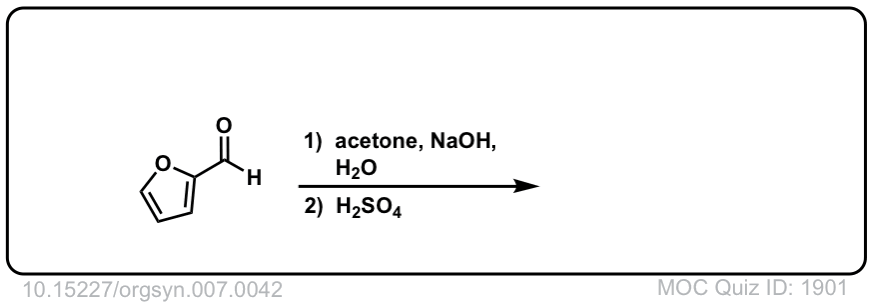 点击翻转
点击翻转
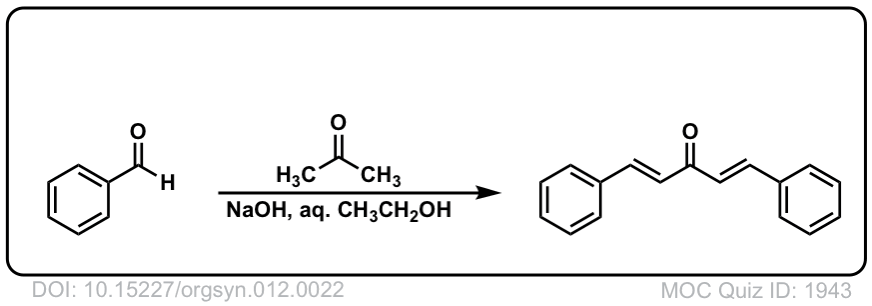
Org。Synth。1932年,12日22
链接DOI: 10.15227 / orgsyn.012.0022
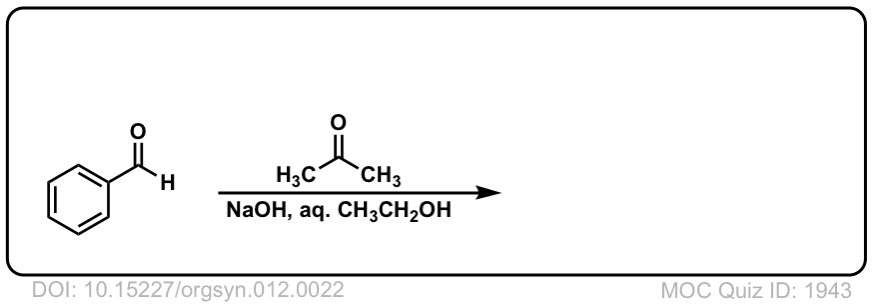 点击翻转
点击翻转
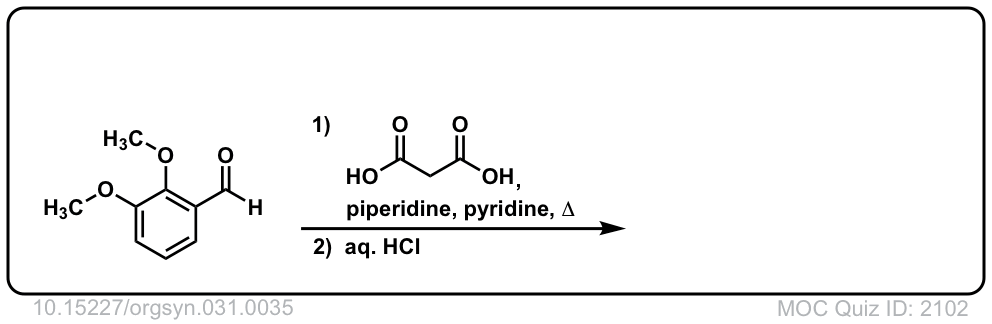
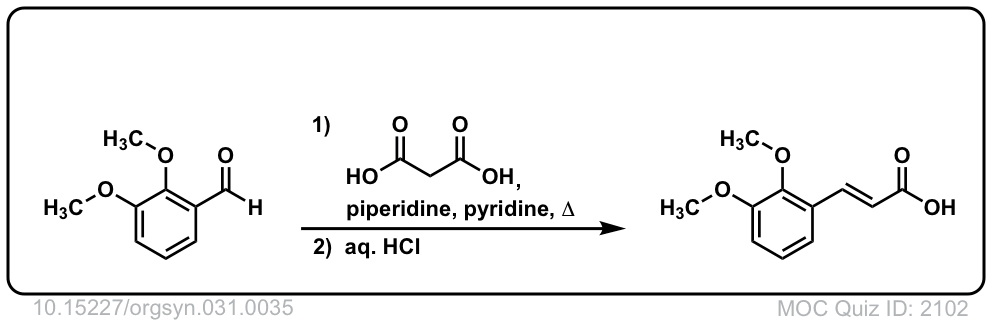
Org。Synth。1951年,31岁,35岁
链接DOI: 10.15227 / orgsyn.031.0035
 点击翻转
点击翻转
非常神奇的
谢谢你注意醛缩酶的8。我很困惑如何1,6-fructose酮糖醇醛因为教科书总是画环形式。很有道理,现在!
高兴你有帮助亚历山大- 99%人不读到笔记,但我感谢小比例(比如你)谁做!
如此强大和丰富!
很高兴你有帮助罗伯特!